
The Bayh-Dole Act’s Vital Importance to the U.S. Life-Sciences Innovation System

Misusing the “march-in right” provision of the Bayh-Dole Act could negatively impact U.S. life-sciences innovation and result in fewer new drugs.
KEY TAKEAWAYS
Key Takeaways
Contents
Complementary Actors in the U.S. Life-Sciences Innovation System. 5
The National Institutes of Health. 5
The Foundation for the National Institutes of Health. 6
The National Center for Advancing Translational Science 7
Small Business Innovation Research Program and Small Business Technology Transfer Program 7
NIH Centers for Accelerated Innovations and Research Evaluation and Commercialization Hubs 8
Public and Private R&D Investments Are Complementary 12
Academic Studies of Public- and Private-Sector Contributions to Drug Discovery and Development 14
Spark Therapeutics/Luxturna 20
The Critical Role of the Bayh-Dole Act 22
The Risks Inappropriate Use of March-In Rights Pose to the Bayh-Dole Act 25
Bayh-Dole March-In Rights Were Never Intended to Address Drug Price Concerns 26
Allowing March-In Rights to Address Pricing Would Lead to Significantly Reduced Innovation. 28
The Bayh-Dole Act and U.S. International Life-Sciences Competitiveness 33
Overview
The United States leads the world in novel biomedical innovation, thanks in large part to its strong research universities, talented researchers, efficient drug approval processes, and a pricing system that enables companies to earn sufficient revenues to reinvest in future generations of innovation. Yet two other factors are indispensable: First, world-leading public and private investment in life-sciences research and development (R&D) activity, with these public and private investments playing unique yet complementary roles in America’s life-sciences innovation system. Second, effective technology transfer and commercialization policies that enable federally funded basic life-sciences research, often taking place at U.S. universities, to lead to research discoveries and inventions that can be patented and licensed to private-sector entities, which then undertake the risky, complex, and expensive process of bringing innovative new medicines and therapies to market. Underpinning this successful ecosystem is bipartisan legislation that gives universities rights to intellectual property (IP) generated from federal funding: the Bayh-Dole Act. Since its introduction in 1980, the act has played a catalytic role in stimulating innovation across many sectors, and especially in the life-sciences. However, calls made by some to use Bayh-Dole “march-in right” provisions to control drug prices threaten to undermine this successful ecosystem and reduce the pace of American biopharmaceutical innovation.
Introduction
The United States leads the world in life-sciences innovation. In the 2000s, more new chemical entities were developed in the United States than in the next five nations—Switzerland, Japan, the United Kingdom, Germany, and France—combined.[1] Broadening the lens to the years 1997 to 2016, U.S.-headquartered enterprises accounted for 42 percent of new chemical and biological entities introduced across the world, far outpacing contributions from European Union member countries, Japan, China, and other nations.[2]
America’s global leadership in life-sciences innovation stems from robust and complementary public and private investments in R&D as well as effective technology-transfer policies that enable private-sector entities to commercialize therapies leveraging scientific discoveries stemming in part from federally funded basic scientific research.
But this has not always been the case. In the latter half of the 1970s, European-headquartered enterprises introduced more than twice as many new drugs to the world as did those in the United States (149 to 66).[3] Throughout the 1980s, fewer than 10 percent of new active substances (i.e., new drugs) were introduced first in the United States, as figure 1 shows.[4] And, as recently as 1990, the global research-based pharmaceutical industry invested 50 percent more in Europe than in the United States.[5] As Shanker Singham of the Institute of Economic Affairs notes, “Europe was the unquestioned center of biopharmaceutical research and development for centuries, challenged only by Japan in the post-war period.”[6]
Figure 1:U.S. Share of New Active Substances (NAS) Launched on the World Market[7]
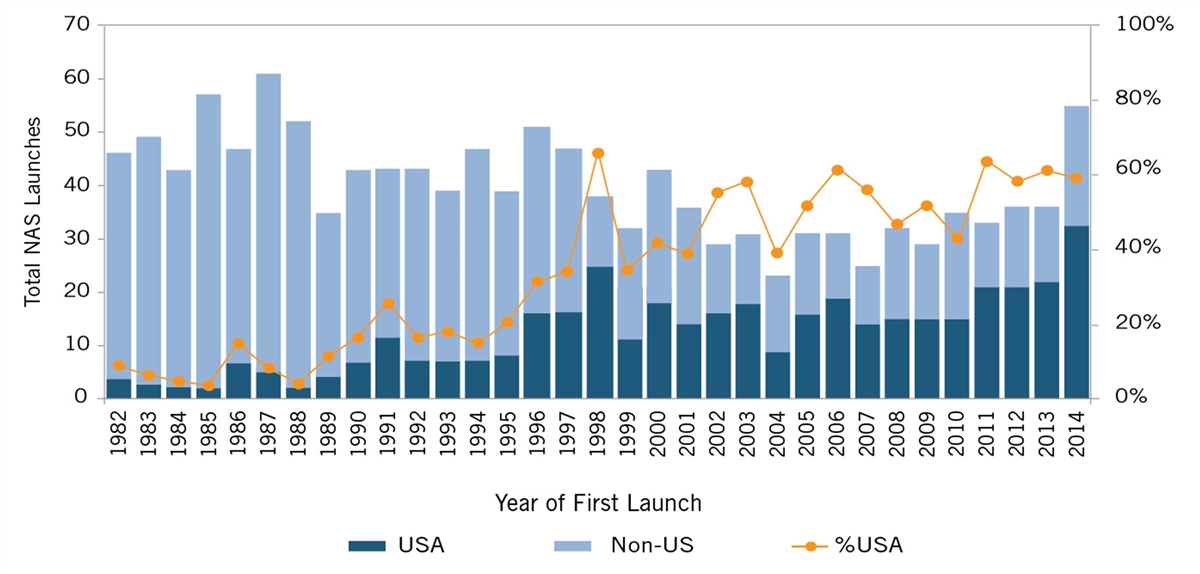
Yet, in recent decades, the picture has changed. Whereas less than 10 percent of new drugs were first introduced in the United States in the 1980s, by the 2010s, more than 60 percent of new drugs were first introduced in the United States.[8] By 2006, pharmaceutical companies invested 40 percent more in the United States than in Europe. And the United States has been the world’s largest funder of biomedical R&D investment over the past two decades, a share some analyses estimate reached as high as 80 percent over that time period.[9] This has contributed to an unprecedented era of life-sciences innovation, and the creation of millions of good U.S. jobs. Over the last decade, biopharmaceutical companies have invested over half a trillion dollars in R&D, while more than 350 new medicines, many firsts of their kind, have been approved by the U.S. Food and Drug Administration (FDA).[10]
America’s wresting of global life-sciences leadership has been no accident, but rather the result of a series of intentional policy decisions designed to make America the world’s preeminent location for life-sciences research, innovation, and product commercialization. For instance, the United States’ introduction of the world’s first R&D tax credit in 1981 played a catalytic role in spurring greater levels of private-sector R&D. In the life-sciences sector, this was complemented by the 1986 introduction of the orphan drug tax credit, which allows drug manufacturers to claim a tax credit on research costs for orphan drugs (i.e., drugs for rare diseases affecting 200,000 or fewer U.S. patients). The 1992 introduction of the bipartisan Prescription Drug User Fee Act (PDUFA), which authorized the FDA to collect user fees associated with applications from the biopharmaceutical industry for regulatory approval of new human-drug submissions, has played a pivotal role in reducing the time it takes the FDA to make safety and efficacy determinations for new drugs—from the over 30 months it took on average in the mid-1980s to less than 10 months today.[11] The United States has also benefitted greatly from having a drug-pricing system that permits companies to earn sufficient revenues from one generation of biomedical innovation to reinvest in the next.[12] That matters greatly because, as the Organization for Economic Cooperation and Development (OECD) has clearly stated, “There exists a high degree of correlation between pharmaceutical sales revenues and R&D expenditures.”[13]
America’s wresting of global life-sciences leadership has been no accident, but rather the result of a series of intentional policy actions.
Increases in federal funding for health research also played a key role. National Institutes of Health (NIH) funding increased from around 0.12 percent of GDP in 1979 to around 0.21 percent in 2009—much of which went to research universities. The combination of increased funding for health research coupled with better policies to help transfer those discoveries to the marketplace played an important role in spurring the growth of the United States’ U.S. biopharmaceutical competitive advantage. In particular, the Bayh-Dole Act, which affords universities rights to the intellectual property generated from federal funding, has played a key enabling role. The act, passed in 1980, permitted contractors, such as universities, small businesses, and nonprofit research institutions, to secure intellectual property rights, often in the form of patents for molecular compounds or biotechnological processes, which in turn may be licensed to private-sector entities, including start-ups and existing biopharmaceutical companies, which then invest the hundreds of millions—often billions—of dollars required to bring safe and effective drugs to market.
Notwithstanding this success, some advocates and elected officials have called for using a provision in the Bayh-Dole Act termed “march-in rights” to allow the government to forcibly license privately owned patents (if the underlying intellectual property can be traced in part to federally funded research) to third parties for the purpose of controlling drug prices. Yet, as this report shows, the congressional architects of the act intended march-in rights to be applied in a very limited set of circumstances, primarily to ensure patent owners were commercializing the subject inventions; they did not intend for march-in rights to be applied for the purpose of controlling drug prices. If the federal government were to apply Bayh-Dole march-in rights for these purposes today, it would threaten to jeopardize America’s successful life-sciences innovation system, as companies would be highly reticent to license IP that could be connected to federal research and subsequently invest the additional billions required to develop a drug if they knew the government could come in as long as two decades later and seize or compulsorily license companies’ IP whenever it deemed a drug’s price too high.
This report begins by providing an overview of key actors in America’s life-sciences innovation system, contextualizing roles played by the federal government and its key research agencies, academic institutions, and the private sector. It then examines the complementary nature of public and private life-sciences R&D investments. The report then uses four case studies to demonstrate how the Bayh-Dole Act works in practice, tracing how new drugs, devices, and biotechnological processes develop—from their earliest roots in federally funded basic research to private-sector development and, finally, to market. In conclusion, the report considers the history and impact of the Bayh-Dole Act, and explains why its march-in rights provision was intended for only a very limited set of cases—not including drug price controls—and that applying march-in rights would have detrimental impacts on both U.S. life-sciences innovation and the development of new drugs.
Complementary Actors in the U.S. Life-Sciences Innovation System
Multiple actors, including the federal government, academia, and industry (including both small biotech start-ups and large multinational corporations) play complementary roles at various phases in the U.S. life-sciences innovation process. While “drug populists” on the left argue that government should primarily take over drug development from the private sector, and “drug libertarians” on the right want to remove the government’s role entirely and have the private sector play the predominant role in drug development, the reality is that the contributions of both are essential.[14]
The National Institutes of Health
The federal government, principally through the National Institutes of Health, funds basic research in the life sciences that sets the stage for the industry-led applied research and development activity that leads to the commercialization of new medicines and treatments.[15] NIH is composed of 27 institutes and centers, each with a specific research agenda, often focusing on particular diseases or body systems.[16] Congress provided NIH a $37.3 billion budget in 2018, and approved legislation raising its funding to $39.1 billion in 2019.[17] More than 80 percent of NIH’s budget funds extramural research through grants, contracts, and other awards, with this research performed by more than 300,000 individuals working at over 2,500 hospitals, medical schools, universities, and other research institutions.[18] The majority of NIH funding is for “basic” research that aims to extend the frontiers of medical understanding, although it’s estimated that approximately one-third of NIH funding supports clinical research (including patient-oriented research, clinical trials, and epidemiological and behavioral studies, as well as outcomes and health services research) that is more applied in nature.[19] NIH also supports training grants that help develop the U.S. scientific and medical workforce. In 2016, NIH grants directly supported the training of more than 9,500 predoctoral students and almost 5,900 postdoctoral fellows.[20]
NIH researchers contribute new-to-the-world life-sciences research. In 2016 alone, over 115,000 articles acknowledged NIH grant support. Moreover, each R01 grant, the most common type of NIH research project grant, leads to an average of 7.36 published research articles. These grants are subsequently cited by other researchers: It’s estimated that each single NIH research grant leads to an average of almost 300 citations in academic literature. The groundbreaking basic life-sciences research supported by NIH is attested to by 153 NIH-supported researchers having won a Nobel Prize.[21]
NIH-funded basic life-sciences research—for instance, into understanding the fundamental processes by which diseases develop and are transmitted, or identifying novel biomarkers that signal the presence of a disease—create a platform for innovation that has led not only to the discovery of new medicines, but to new tests (e.g., blood tests for substances), new procedures (e.g., improved cardiac stents that substitute for surgery), and new equipment (e.g., gene sequencers).[22] NIH-funded research has supported discoveries that have contributed to reduced deaths from cancer and lower rates of disability due to stroke, heart disease, hepatitis B, and osteoporotic fractures.[23] NIH-supported research has also led to the development of anti-AIDS drugs; the discovery of neurotransmitters, which led to the development of anti-depression treatments leveraging selective serotonin reuptake inhibitors (SSRIs); and treatments reducing scar tissue formation.[24] Federally funded research has contributed to breakthrough therapies, including the anticancer drug imatinib (Gleevec), tumor necrosis factor blockers such as infliximab (Remicade) and etanercept useful in inflammatory rheumatologic and gastroenterological diseases (Enbrel), and vascular endothelial growth factor inhibitors such as bevacizumab (Avastin) for cancer and eye diseases.[25]
While NIH is a sprawling enterprise with 27 institutes and centers each playing an important role in life-sciences innovation, the unique role of several particular units is worth highlighting.
The Foundation for the National Institutes of Health
The Foundation for the National Institutes of Health (FNIH) is a 501(c) (3) charitable organization chartered by Congress in 1996 that procures funding and manages alliances with public and private institutions in support of NIH’s mission.[26] The foundation is legally chartered to accept donations from alumni inventors and scientists, philanthropists, and high-wealth individuals to support activities designed to accelerate biomedical research and strategies to fight against diseases in the United States and across the world. FNIH organizes and administers research projects; supports education and training of new researchers; organizes educational events and symposia; and administers a series of funds supporting a wide range of health issues. Since its founding, it has raised over $1 billion (over $80 for each $1 of NIH funding it has received) from 9,200 donors, which it has used to support over 600 research programs.[27] FNIH specializes in building public-private partnerships between government, academic, industry, nonprofit, and patient-group researchers in order to conduct research into specific disease states and research areas. For instance, FNIH has played a key role in spearheading the Biomarkers Consortium, a collaboration that brings researchers from a variety of fields together to rapidly identify, develop, and qualify biomarkers (a biological indicator of a disease state in the body) with the goal of advancing new therapies and guiding improvements in regulatory and clinical decision-making.[28] Institutions such as FNIH matter because partnerships have become increasingly important in life-sciences innovation. In fact, from 2005 to 2014, the number of early-stage R&D partnerships more than doubled, from 256 to 578; the number of life-sciences R&D partnerships also more than doubled, from 4,000 to 9,000; and the number of consortia increased almost tenfold, from 34 to 334.[29] FNIH is an important convener and facilitator in America’s life-sciences innovation system.
The National Center for Advancing Translational Science
Established by Congress in December of 2011, the National Center for Advancing Translational Sciences (NCATS) focuses on ways to reduce, remove, or bypass system-wide bottlenecks in the translational process: the process of turning observations in the laboratory, clinic, and community into interventions that improve the health of individuals and the public—from diagnostics and therapeutics to medical procedures and behavioral changes.[30] The core goal of NCATS is to help get more treatments to more patients more quickly. NCATS focuses not on specific diseases, but on what is common among them and in the translational science process. Essentially, NCATS studies translation on a system-wide level as a scientific and operational problem.[31] NCATS initiatives focus on topics such as discovering new therapeutic uses for existing molecules, improving the availability of rare disease information, treatment, clinical studies, and general awareness for both patients and the medical community via programs such as the Rare Diseases Clinical Research Network (RDCRN), tissue chips for drug screening, and toxicology in the 21st century.[32]
Small Business Innovation Research Program and Small Business Technology Transfer Program
The Small Business Innovation Research (SBIR) and Small Business Technology Transfer (STTR) programs, collectively known as America’s Seed Fund, represent one of the largest sources of early-stage capital for technology commercialization in the United States, allowing U.S.-owned and operated small businesses to engage in federal research and development that has a strong potential for commercialization.[33] NIH’s SBIR program funds early-stage small businesses that are seeking to commercialize innovative biomedical technologies; STTR is similar but requires small businesses to engage with a research institution. Eleven federal agencies operate SBIR/STTR programs. Of the institutes or centers within NIH, 24 have SBIR and STTR programs. In 2017, NIH’s SBIR and STTR programs invested almost $1 billion ($861 million for SBIR; $121 for STTR) into promising, young, innovative life-sciences start-ups. In 2017, NIH funded 1,520 start-ups. For FY 2019, NIH will disburse $1 billion in SBIR funds and $141 million for STTR. Funds will be disbursed over a series of two phases: Phase 1 feasibility studies (grants of up to $150,000), which may be extended into Phase II development activities funded at $1 million, with a possibility of a Phase IIB competing renewal award.[34] SBIR plays a key role in America’s innovation system, and particularly in the life-sciences sector. A number of groundbreaking life-sciences companies got a kick start from SBIR, including Genzyme (biotech therapies), Affymetrix (GeneChip), Amgen (biopharmaceuticals), Jarvik Heart (artificial heart), Biogen (neurological, autoimmune therapies), Millennium Pharma (gene databases), Geron (telomerase inhibitors for cancer treatment), and Neocrine Bioscience (neurological and endocrine pharmaceuticals).[35] SBIR plays a major role in making projects that would not happen otherwise possible. For instance, a study of NSF SBIR Phase II recipients found that 75 percent thought their project probably or definitely would not have proceeded absent program funding: 34 percent were definite and 41 percent thought it unlikely.[36] In short, NIH’s SBIR/STTR program represents an indispensable asset within America’s life-sciences innovation system.
The federal government primarily funds basic scientific research, while private-sector companies perform much of the applied R&D, including the completion of clinical trials required to transform basic scientific research into commercial products.
NIH Centers for Accelerated Innovations and Research Evaluation and Commercialization Hubs
The NIH Centers for Accelerated Innovations (NCAI) and the NIH Research Evaluation and Commercialization Hubs (REACH) are focused on accelerating the translation of scientific discoveries into commercial products. The programs represent public-private partnerships with expertise and resources from the federal government, academia, and the private sector that will change the way discoveries with scientific and commercial potential are identified and developed. The National Heart, Lung, and Blood Institute started NCAI in September 2013 and is the primary federal supporter of the program, merging the strengths of 14 high-impact research institutions.[37]
REACH provides qualified institutions with the initial investment and resources to nurture innovators to identify and develop high-priority early-stage technologies related to NIH’s objectives by providing: infrastructure for identifying the most promising technologies; funding for product-definition studies (e.g., feasibility studies, prototype development, and proof-of-concept studies); coordinated access to expertise in areas required for early-stage technology development (including scientific, regulatory, reimbursement, business, legal, and project management); and skills development and hands-on experience in entrepreneurship. There are three NIH Research Evaluation and Commercialization Hubs: the Long Island Bioscience Hub, ExCITE, and MN-REACH. Congress chartered the hubs as part of the 2011 SBIR/STTR reauthorization, with the vision that the hubs would provide Phase 0 proof of concept partnerships to help get more promising start-ups into the SBIR program.
The Life-Sciences Industry
Private-sector life-sciences enterprises undertake the difficult, risky, and expensive process of conducting the applied research and development activities—everything from synthesizing molecular compounds to conducting clinical trials proving the safety and efficacy of new medicines—required to turn basic life-sciences discoveries into innovative medicines, therapies, or devices brought to market. Essentially, the private sector leads the translation of basic research findings into new medicines.
The life-sciences industry is perhaps the world’s most R&D intensive, investing 20.4 percent of its sales into R&D globally.[38] In the United States, the life-sciences sector invests over 21 percent of its sales into R&D, while accounting for 23 percent of domestic R&D funded by U.S. businesses—more than any other sector.[39] In 2013, America’s life-sciences companies invested $96.5 billion in R&D, of which $74.5 billion was self-funded and 84 percent occurred in the United States.[40] Measured by R&D expenditure per employee, the U.S. biopharmaceutical sector leads all other U.S. manufacturing sectors, investing more than 10 times the amount of R&D per employee than the average U.S. manufacturing sector.[41] Globally, the pharmaceutical industry invested over $1.36 trillion in R&D in the decade from 2007 to 2016.[42] Global annual private-sector R&D investment reached $156.7 billion in 2016, and is expected to grow to $181 billion annually by 2022.
The industry must be so R&D-intensive because bringing innovative new drugs to market is risky, time-consuming, and expensive. Figure 2 shows the rigorous series of stages involved in bringing a new drug to market, beginning with basic research, drug discovery, and preclinical trials; then proceeding to three stages of human clinical trials, which culminate in a drug’s approval (or rejection) by the FDA; followed by pharmacovigilance (or post-approval safety monitoring). Biopharmaceutical companies conduct laboratory screening of 5,000 to 10,000 chemical compounds for each new drug approved for use in humans. On average, of the 5,000 to 10,000 compounds that are screened, approximately 250 enter preclinical testing, and 5 enter clinical testing.[43] Moreover, less than 12 percent of candidate medicines that even make it into Phase I clinical trials are ultimately approved by the FDA.[44]
Figure 2: The R&D Process for New Drugs[45]
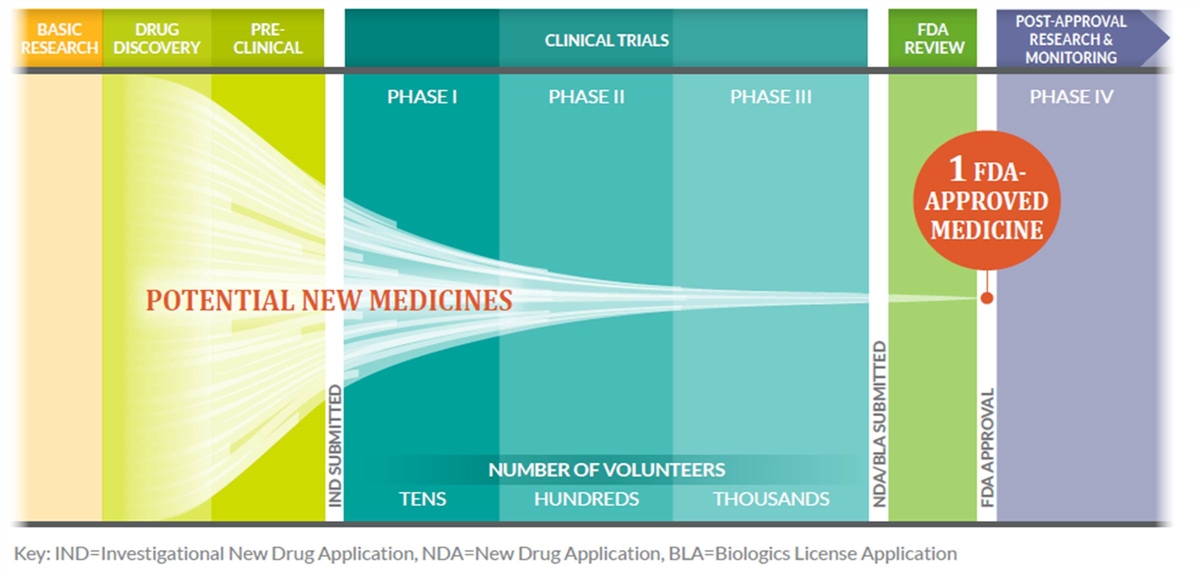
This process is expensive. The development of new drugs requires years of painstaking, risky, and expensive research that, for a new pharmaceutical compound, takes an average of 11.5 to 15 years of research, development, and clinical trials, at a cost of $1.7 to $3.2 billion.[46] For instance, one in-depth study conducted by the Tufts University Center for the Study of Drug Discovery, “Cost of Developing a New Drug,” estimated that the average cost of developing a new drug in 2014 was $2.56 billion. This estimate was based on the costs of 106 investigational new drugs and biologics, from 10 firms, first tested on humans between 1995 and 2007. It included out-of-pocket clinical costs, discovery research expenses, and preclinical development costs—plus the cost of capital and the cost of developing drugs that did not make it into the marketplace.[47] And while, as noted, only a tiny fraction of drugs that enter clinical trial testing are ultimately approved by the FDA, an even smaller fraction of approved drugs ever becomes economically profitable. A study released in 2010 by Vernon, Golec, and DiMasi found that 80 percent of new drugs made less than their capitalized R&D costs. Entities in the second-most-profitable decile barely broke even; those in the first decile had discounted profits that were more than twice their discounted R&D costs.[48] Other studies have found that of the most successful 10 percent of approved drugs, only 1 percent of those that entered clinical trials—maybe three new drugs each year—generate half of the profits of the entire drug industry.[49]
In 2018, the Congressional Budget Office (CBO) estimated that because of the high failure rate, biopharmaceutical companies would need to earn a 61.8 percent rate of return on their successful new drug R&D projects in order to match a 4.8 percent after-tax rate of return on their investments (i.e., a risk-free rate they could readily attain in public markets).[50] The 61.8 percent figure is driven by two assumptions. The first is that 90 percent of new biopharmaceutical research and development projects fail, meaning all profits must come from the 10 percent that succeed. The second assumption in the CBO study was that it takes 12 years—during which companies spend large sums on development, testing, and approvals—for a successful project to start earning revenues. Concentrating only on the rate of return to successful projects therefore gives a misleading picture of the overall profitability of biopharmaceutical companies.
One study finds that life-sciences companies invest over $100 in development for every $1 the government invests in research leading to an innovation.
Despite these challenges, the industry continues to bring innovative new drugs to market. Biopharmaceutical companies have about 7,000 medicines under development globally, with 4,000 of them in the United States.[51] This includes more than 1,100 cancer-treating drugs, 566 of which target rare diseases, 537 treat neurological disorders, over 300 target autoimmune diseases, 200 target heart diseases and strokes, 170 are for diabetes, and 85 target Alzheimer’s.[52] It’s estimated that 74 percent of drugs in the clinical pipeline are potential first-in-class medicines (meaning they represent a possible new pharmacological class for treating a medical condition), including 86 percent for the treatment of Alzheimer’s, 79 percent for cancer, 75 percent for psychiatric treatments, and 73 percent of drugs targeting cardiovascular- or diabetes-related illnesses.[53]
The industry also drives U.S. global competitiveness. The pharmaceutical and medical-instrument subsectors’ combined output reached $675 billion in 2015 (almost 4 percent of U.S. GDP). In 2016, the sector supported nearly 5 million workers, including employing 1.2 million workers directly (with jobs paying an average annual wage of $124,400) and creating an additional 3.5 million jobs indirectly.[54]
America’s life-sciences innovation sector is comprised of a mix of large established companies and small firms (often start-ups). This matters because, as a recent ITIF study found, high-technology start-up companies account for a significant share of new-employment growth, and a higher portion of job growth than start-ups in other industries.[55] Indeed, an estimated two-thirds of U.S. pharmaceutical firms are start-up companies.[56] These companies are extremely R&D intensive. For example, the average biotech firm invests 20 percent of its revenues on R&D, while start-ups invest 62 percent.[57] Most importantly, small firms account for more than half of the new drugs created in the United States.[58]
Universities
Universities play a key role in America’s life-sciences innovation system. In 2017, U.S. universities conducted $68.2 billion in research activity, with $39.8 billion funded by federal sources; $23.6 billion contributed by “other” sources, including universities themselves and other nonprofit research institutions; and $4.8 billion contributed by industry.[59] In 2015, when U.S. universities conducted $68.8 billion on R&D, 56.5 percent was dedicated to life-sciences-oriented research.[60] In 2015, U.S. universities themselves invested $8.6 billion in medical and health research, which accounted for 5.5 percent of all U.S. investment in the field that year.[61]
Dr. Robert Kneller, a professor at the University of Tokyo’s Research Center for Advanced Science and Technology, has found that the universities supported by NIH play a more specialized and advanced role in the overall process of medical innovation.[62] Kneller discovered that NIH-funded entities, including universities, generally produce new pharmaceutical discoveries that are more advanced, more likely to be considered “novel” (as opposed to advances that are based on a preexisting substance that is modified and resubmitted for approval), and more likely to be orphan drugs (substances that address rare diseases or conditions).[63]
Similarly, Sampat and Lichtenberg, in a study of 478 drugs approved by the FDA from 1998 to 2005, found that 12.7 percent had an underlying academic patent, including 5.8 percent of standard-review drugs and 22.6 percent of priority-review drugs. Their study found even larger indirect effects on the innovation process, with 23.7 percent of standard-review drugs, 45.8 percent of priority-review drugs, and 32.7 percent of all new drugs citing an academic patent.[64] In another analysis, Lowe found that about 15 percent of new drug discoveries come from academic labs.[65]
Universities play a key role in America’s innovation system, especially in the life-sciences.
Industry-academic partnerships also play an important and growing role. In 2017, biopharma companies provided over $2.5 billion in research funding to America’s universities—in all 50 states—accounting for more than 60 percent of industry funding of university research.[66] There exist a number of examples of companies funding university research—as drug innovation relies so heavily on scientific breakthroughs. For instance, companies such as Amgen, GlaxoSmithKline, Novartis, and Vertex have funded research and clinical trials at Duke University.[67] AbbVie has entered into a partnership with the University of Chicago for cancer research.[68] Astellas provided $26 million to the M.D. Anderson Cancer Center in Houston to support treatment for acute myeloid leukemia.[69] Novartis has supported more than 300 academic collaborations, such as with Harvard University on the Zika virus.[70] Pfizer established its Global Centers for Therapeutic Innovation in an $85 million partnership with the University of California at San Francisco.[71]
Public and Private R&D Investments Are Complementary
Public and private R&D investments and activities are highly complementary—and indispensable—to America’s successful life-sciences innovation system.
Complementary Roles
Historically, public-sector researchers have performed the upstream, earlier-stage research elucidating the underlying mechanisms of disease and identifying promising points of intervention, whereas corporate researchers have performed the downstream, applied research resulting in the discovery of drugs for the treatment of diseases and have carried out the development activities necessary to being them to market.[72] Federally funded basic life-sciences research tends to be concentrated in the basic science of disease biology, biochemistry, and disease processes, with a major goal of the research being the identification of biomarkers and biologic targets that new drugs could treat.[73] While the private sector does invest in basic scientific research, including at U.S. universities, the preponderance of its activity is applied R&D focused on the discovery, synthesis, testing, and manufacturing of candidate compounds intended to exploit biologic targets, for the purpose of curing medical conditions.[74]
A number of studies have elucidated this dynamic. For instance, a 2000 study by the U.S. Senate Joint Economic Committee found that, “Federal research and private research in medicine are complementary. As medical knowledge grows, federal research and private research are becoming more intertwined, building the networks of knowledge that are important for generating new discoveries and applications.”[75] Similarly, as an OECD study has argued, “It is particularly important for government-funded research to continue to provide the early seeds of innovation. The shortening of private-sector product and R&D cycles carries the risk of under-investment in scientific research and long-term technologies with broad applications.”[76]
Public and private R&D investments and activities are highly complementary—and indispensable—to America’s successful life-sciences innovation system.
Government funding of early-stage research is critical. As America’s National Academy of Sciences wrote, “Fewer investments in basic research (by NIH) can result in fewer new drug therapy candidates, which in turn can result in fewer investments by private industry to advance promising candidates.”[77] Likewise, the Tufts Center for Drug Development concluded, “Scientific and development histories demonstrate the rich interconnectivity of all sectors in the drug-discovery and drug-development ecosystem.”[78] This affirms a 2015 Battelle Memorial Institute study that found, “NIH funded research produced an average of 5.9 patents per $100 million in R&D expenditures from 2000–2013—or at a rate of one patent per every $16.9 million in NIH funding.”[79] These findings are similar to those from a 2017 National Bureau of Economic Research study which found that, “An additional $10 million in NIH funding for a research area generates 2.3 additional private-sector patents in that area, or roughly 1 patent for every 2 to 3 NIH grants.”[80] Similarly, the 2015 Battelle report went on to find that, “NIH patents also averaged 5.14 forward citations, meaning the NIH is an integral part of the knowledge chain for $105.9 million in downstream R&D for every $100 million in taxpayer funded awards. These downstream connections represent other research organizations, in both the private and public sector, leveraging NIH discoveries into follow-on R&D spending.”[81]
One reason federal support for basic and early-stage applied research is a complement to private-sector research is that industry is able to build on the knowledge discoveries from publicly supported life-sciences research, making their own research more productive and effective. Public funds are so important to the life-sciences industry because, unlike for many other industries, the majority of value added from the life-sciences industry is derived from radical innovation drawn from basic science. At the advent of the modern life-sciences industry in the 1960s and 1970s, it typically took many years for federally funded research to impact the private sector. More recent revolutions in techniques in biotechnology, including “mechanism-driven” drug design, have made publicly funded basic R&D more relevant to pharmaceutical firms in the near term.[82] Indeed, the lifespan of R&D to commercialization (as defined by patents) is substantially longer in the life sciences than in other industries, taking an average of between 12 and 14 years.[83] In this sense, publicly funded basic R&D generates more than just papers, pure knowledge, and postgraduates; public-sector funds increase the productivity of the industry as a whole by facilitating an environment of readily valuable basic R&D. Public R&D within the life-sciences industry leads to the development of “infrastructure knowledge,” or skills acquisition, techniques, and research tools that increase the expected rate of return for private-sector R&D projects.[84]
These “spillovers” effectively provide firms with a common platform of basic knowledge, and thus induce greater levels of innovation. For the life-sciences industry, Dr. Everett Ehrlich found that a dollar of NIH support for research leads to an increase of private medical research of roughly 32 cents.[85] After reviewing over 60 academic articles on the relationship between public- and private -sector R&D, Cockburn and Henderson concluded:
There are a number of econometric studies that, while imperfect and undoubtedly subject to improvement and revision, between them make a quite convincing case for a high rate of return to public science in this [life-sciences] industry. It is worth noting that there are, so far as we are aware, no systematic quantitative studies that have found a negative impact of public science.[86]
The aforementioned 2017 NBER study examined whether there was evidence of NIH investments either “crowding out” or “crowding in” private-sector investment. As the authors wrote, their findings were “consistent with the absence of crowd out” and “suggest that NIH funding spurs private patenting by either increasing total firm R&D expenditure or increasing the efficiency of these expenditures.”[87] Additionally, “Even if NIH funding crowds out some private investment, it is offset by increases in the number of patents related to NIH funding through indirect citation channels, or by increases in the productivity of private R&D investments.”[88]
Publicly funded basic R&D generates more than just papers, pure knowledge, and postgraduates; public-sector funds increase the productivity of the industry as a whole by facilitating an environment of readily valuable basic R&D.
Similar findings were reported in a 2012 Milken Institute study, which found that $1 of NIH funding boosted the size of the bioscience industry by $1.70 and that the long-term impact may be as high as $3.20 for every dollar spent.[89] Likewise, a 2013 report by Battelle found that, looking solely at federal support for the Human Genome Project between 1988 and 2012, every dollar of federal funding helped generate an additional $65 dollars in genetics-related private activity.[90] Rutgers University Professor A.A. Toole identified a quantifiable correlation between investment in publicly funded basic research and corporate-funded applied research wherein an increase of 1 percent in the funding of public basic research led to an increase of 1.8 percent in the number of successful applications for new molecular entities (compounds that have not been approved for marketing in the United States) after a lag of about 17 years. Toole concluded that a $1 investment in public-sector basic research yielded $0.43 in annual benefits in the development of new molecular entities in perpetuity—a remarkable return on investment.[91] Similarly, Lichtenberg estimated a social return from pharmaceutical innovation of 67.5 percent.[92] The total social return from biomedical research (public and private) has been estimated at 150 percent, implying that society would benefit from a significant increase in research spending (which is ironically the opposite of what would likely happen if there were widespread restrictions on drug prices.)[93]
However, it’s critical to remember that considerable investment is required to bring a drug to market even after considerable amounts of basic research have been conducted. In fact, one study found that biotechnology companies invest $100 in development for every $1 the government invests in research that leads to an innovation.[94]
Academic Studies of Public- and Private-Sector Contributions to Drug Discovery and Development
A variety of academic studies have examined the contributions made by public, university, and private-sector entities toward the development of new drugs. For instance, a study of 32 innovative drugs introduced before 1990 found that without the contributions of government laboratories and other noncommercial institutions, approximately 60 percent of the drugs would not have been discovered or would have had their discoveries markedly delayed.[95] Likewise, a 2011 study by Stevens et al. found that over the past 40 years, 153 new FDA-approved drugs, vaccines, or new indications for existing drugs were discovered through research carried out in public-sector research institutions (PSRIs).[96] Stevens et al. argued that, “PSRIs have contributed to the discovery of 9.3 to 21.2 percent of all drugs involved in new-drug applications approved during the period from 1990 through 2007.”[97] However, Thomas Sullivan noted the flip side of the Stevens et al. study, observing that “while the authors identified 153 FDA-approved drugs that were discovered at least in part by PSRIs during the past 40 years, they fail to mention the number of FDA-approved drugs during that same period discovered by industry, likely because that includes 80–90 percent of all drugs.”[98]
In a 2011 study, Sampat and Lichtenberg built a database of 478 new drugs, including 379 approved by the FDA between 1998 and 2005 that had direct or indirect public support, where a direct role involved, for instance, a government entity (i.e., a federal agency) securing a patent, and an indirect role including, for example, government funding of basic underlying research that is built upon in the drug discovery process.[99] The study found that 9 percent of approved drugs had a public-sector patent, 25.3 percent cited at least one public-sector patent, 41.4 percent cited at least one government publication, and 47.8 percent cited either a public-sector patent or a government application. The authors found that “the indirect influence of the public sector on drug development was much larger than the direct effect.”[100]
Other studies have emphasized the importance of private-sector contributions in drug discovery and development. For instance, Zycher, DiMasi, and Milne identified 35 drugs and drug classes (a group of drugs used to treat a given medical condition in similar ways), finding that “the scientific contributions of the private sector were crucial for the discovery and/or development of virtually all of the thirty-five drugs or drug classes.”[101] Specifically, they found that among 35 drugs and drug classes, private-sector research was responsible for central advances in basic science for 7, in applied science for 34, and in the development of drugs yielding improved clinical performance or manufacturing processes for 28.[102] As the authors wrote, “Without the scientific advances yielded by private-sector research, most drugs would not be developed, and thus the economic returns to publicly funded research would be sharply reduced.”[103]
Updating this work in 2016, DiMasi, Milne, Cotter, and Chakravarthy examined the roles of the private and public sectors in drug development by examining an array of evidentiary materials on the history of 19 individual drugs, 6 drug classes, and 1 drug combination identified as the most transformative drugs in health care over the past 25 years by a survey of over 200 physicians.[104] The authors found that only four of the drugs appeared to have been almost completely researched and developed by one sector, although one sector or the other did dominate particular phases of the R&D continuum. Specifically, they discovered 54 percent of basic science milestones were achieved by the public sector and 27 percent by the private sector. The private sector contributed 58 percent of discovery-oriented milestones, while 15 percent were contributed by the public sector. The private sector proved the dominant player in achieving major milestones related to the production and drug development phases (81 percent and 73 percent of the drugs reviewed, respectively).[105] As the authors concluded from their analysis, “Industry’s contributions to the R&D of innovative drugs go beyond development and marketing and include basic and applied science, discovery technologies, and manufacturing protocols, and that without private investment in the applied sciences there would be no return on public investment in basic science.”[106] However, once again, the study points to the complementarity of public and private contributions to life-sciences innovation that have made the U.S. system so successful.
Case Studies
The following section provides several case studies illustrating the complementary roles of key actors in America’s life-sciences innovation system in brining novel medicines, devices, and therapies to market.
Yervov/Ipilimumab
Melanoma is a skin cancer caused by an uncontrolled growth in pigment-producing skin cells. Although it accounts for less than 5 percent of skin cancer cases, melanoma is the most dangerous form of skin cancer, accounting for the vast majority of skin cancer deaths, with 76,250 Americans being diagnosed in 2012 and 9,180 perishing from the disease.[107] Yervoy, a biologic medicine developed in collaboration between Medarex and Bristol-Myers Squibb, is the first drug ever demonstrated to significantly improve overall survival rates for patients with advanced metastatic melanoma, with 25 percent of patients treated with Yervoy still living over four and a half years later.[108]
Federal funding provided by the National Institutes of Health in the 1990s supported basic research on the role of T cells, a subtype of white blood cells that play a key role in the immune system.[109] Dr. James Allison, then a professor in the Division of Immunology, and director of the Cancer Research Laboratory, at the University of California at Berkeley, was a research pioneer who dreamed of harnessing the power of the human immune system to attack cancer. In particular, he sought to understand the body’s immune response to cancer, and how the disease proliferates by suppressing T cells. In healthy individuals’ immune systems, foreign bacteria and viruses—as well as transformed cancer cells—bear molecular structures called antigens that identify them to the immune system as dangerous.[110] T cells attack antigen-bearing targets, but only when the immune system signals to do so—so the T cells do not attack healthy cells—with the immune system using a series of checkpoints that operate like traffic lights, sending signals that either activate or inhibit T cells.[111]
Dr. Allison believed that cancers might work by suppressing T cell activation. In 1995, he demonstrated that a checkpoint molecule called cytotoxic T lymphocyte antigen-4 (CTLA-4) put the brakes on T cell responses. Dr. Allison recognized that if he could block CTLA-4, the body’s immune system could be activated to unleash an antitumor response.[112] In subsequent preclinical experiments, Dr. Allison demonstrated that he could bind a type of protein called a monoclonal antibody to CTLA-4, thereby preventing it from interfering with T cell activation. He spent several years amassing data in mice to show that anti-CTLA-4 antibodies work, before turning his attention to work on developing an antibody for humans.[113] The approach of blocking CTLA-4 from interfering with checkpoint activation (the so-called “checkpoint blockade”) gave rise to the field of cancer immunotherapy, a fundamentally new strategy for treating malignancies that unleashes the immune system to kill cancer cells.[114] Dr. Allison worked with the University of California (UC) Berkeley’s technology transfer office to patent his method for activation of T cells through a blockade of CTLA-4 signaling, receiving a patent from the U.S. Patent and Trademark Office for the “blockade of T lymphocyte down-regulation associated with CTLA-4 signaling.”[115] The patent, assigned to the University of California, explicitly stated the invention was made with government support under contracts CA 40041 and CA 09179 awarded by the National Institutes of Health.[116]
But the process of turning Dr. Allison’s discovery into a commercial medicine that would benefit patients was just getting started. Originally, UC Berkeley licensed the intellectual property to the start-up NeXstar Pharmaceuticals, which subsequently merged with Gilead Sciences, Inc. Gilead then sublicensed the rights to Medarex, which developed a human monoclonal antibody, ipilimumab, and began testing it in partnership with Bristol-Myers Squibb (BSM). BSM acquired Medarex in 2009, taking over responsibility for the clinical development of ipilimumab.[117] By 2010, results from the first Phase III clinical trials demonstrated the drug significantly prolonged the lives of patients with metastatic melanoma.[118] Ipilimumab, which Bristol Myers Squibb would bring to market under the brand name Yervoy, was fast-tracked and approved by the U.S. Food and Drug Administration in March of 2011. Since then, more than 10,000 cancer patients have received Yervoy to treat advanced melanoma and other types of cancer.[119] In October 2018, Dr. Allison was awarded the 2018 Nobel Prize in Physiology or Medicine for his groundbreaking work on cancer immunotherapy.[120]
While Yervoy is saving lives of melanoma patients today, it began with NIH-funded basic researchers who were motivated by a fundamental curiosity about how the human immune system works.
While Yervoy is saving lives today, it began with NIH-funded basic researchers who were motivated by a fundamental curiosity about how the human immune system works.[121] Indeed, Dr. Allison’s original interest in CTLA-4 was driven by his basic curiosity about how T cells are activated and regulated.[122] The Yervoy case also highlights the important role Bayh-Dole plays in supporting university technology transfer and licensing, and also the subsequent challenge of commercialization. As Carol Mimura, UC Berkeley’s assistant vice chancellor, IP and Industry Research Alliances, explained, “Two startup companies took the risk of performing lengthy and expensive R&D for more than a decade on an unproven opportunity.”[123] It was, “entrepreneurship and dogged determination [that] resulted in a product that the pharmaceutical industry was able to take to the finish line.”[124] For its part, UC Berkeley received an upfront payment of $87.5 million when it licensed the rights, which it used to finance other scientific research needs, from faculty retention, new biology teaching labs, and equipment for the cancer research lab to a new building devoted to stem cell research.[125] The Yervoy case study effectively highlights the roles played by key actors across America’s life-sciences innovation system, from the public to universities to the private sector.
CardioMEMS
For patients suffering from heart disease or failure, CardioMEMS provides a remote, pulmonary artery pressure (PAP) monitoring system that helps patients and their doctors to monitor their heart health.[126] CardioMEMS is a monitoring system implanted in a patient’s pulmonary artery that uses a sensor no larger than a dime with two thin loops at each end to measure their pulmonary arterial pressure and heart rate. The system comes with an electronic unit patients use to take daily measurements and then send the results wirelessly to their doctor.
Figure 3: The CardioMEMS Device[127]

Clinical studies have shown that patients whose clinicians use a pulmonary artery sensor to manage heart failure experience fewer hospitalizations and enjoy an improved quality of life.[128] One clinical trial of the device demonstrated a 37 percent reduction in heart failure hospitalizations over a 15 month average follow-up timeframe when physicians used the device to guide patient care.[129] Some 6,000 U.S. heart failure patients have now had a CardioMEMS PAP monitor implanted.[130] However, there remain tens of thousands more U.S. heart failure patients with a recent history of at least one heart failure hospitalization who would be eligible to receive an implanted PAP monitor.[131] Going forward, the technology may be useful for additional applications, including monitoring chronic diseases that have a hemodynamic progression, such as hypertension, pulmonary arterial hypertension, respiratory disease, portal hypertension, and applications within congenital heart disease.[132]
The CardioMEMS is delivering life-changing results for patients suffering from heart failure today, but its history stretches back over 25 years—and provides a compelling example of the roles played by a variety of key players in the U.S. life-sciences innovation system. In the mid-1990s, the Army Research Office sponsored a program through the Multidisciplinary University Research Initiative (MURI) on “Intelligent Turbine Engines.” Mark Allen, an engineering professor at Georgia Tech University, applied for and won a $500,000 grant from the Army Research Office to develop a pressure sensor that could be used in engines to provide control signals to ensure optimal engine performance.[133] He and a colleague designed, fabricated, and tested a new type of pressure sensor that was: small; capable of operating in harsh environments, such as at high temperature; and capable of wireless interrogation. The sensor proved successful. It was highlighted in journal publications and adopted by the Army—and Georgia Tech, leveraging the Bayh-Dole Act, secured several patents for it.[134]
In 2000, with the help of a physician, Professor Allen began to explore how microelectromechanical systems (MEMS)-based manufacturing technologies could be used to create a new generation of medical devices. In particular, they were interested in how wireless sensors could detect disease states from within the body, and recognized that “the turbine engine sensor developed for harsh environments under the MURI research program might also be applicable in another harsh environment, the human body.”[135] In 2001, they incorporated CardioMEMS, licensing key patents from Georgia Tech, to explore commercialization of the technology, starting with wireless sensors as monitors of endovascularly repaired abdominal aortic aneurysms.[136] As Professor Allen noted, “Having clear access to the intellectual property developed in the academic laboratory through the mechanisms of the Bayh-Dole Act was the prerequisite for CardioMEMS’ success.”[137]
The FDA approved the CardioMEMS device in 2005. By 2007, it had been implanted in over 100 patients, and attracted more than $50 million in private equity investment—a ratio of approximately $100 of private investment for each $1 of government investment from the original Army Research Office grant.[138] In June 2014, after St. Jude Medical acquired CardioMEMS, its CEO Daniel Starks commented that the CardioMEMS HF System “sets a new treatment paradigm for heart failure that reduces hospitalizations and improves the quality of life for patients.”[139] In January 2017, Abbott Labs purchased St. Jude Medical, bringing the CardioMEMS into Abbott’s product portfolio.[140] The story of CardioMEMS is a strong example of how federal funding for basic research supports university discoveries that can then get commercialized through spin-outs or licensing to bring innovative products to market that improve quality of life for U.S. (and global) citizens.
Gleevec/Imatinib
Gleevec is a landmark drug for patients suffering from chronic myelogenous leukemia (CML), a bone marrow cancer which results in the excessive production of certain types of white blood cells that proliferate to the point of overwhelming the body’s immune system. CML is the result of a genetic mutation in which there is an improper translocation of chromosomes 9 and 22, resulting in a shorter-than-normal chromosome 22 (the so-called “Philadelphia chromosome”) which harbors a cancer-causing gene, or oncogene, identified as BCR-ABL.[141] The oncogene causes its damage because it represents an “on” switch for cell division that does not have a corresponding “off” position.[142] Gleevec works by jamming BCR-ABL in the “off” position, and in so doing became the first of a new class of drugs called signal transduction inhibitors (STIs), which work by interfering with the pathways that signal the growth of cancerous cells.
The basic science that established cancers as having genetic causes, and that the genetic causes of ostensibly the same cancers in different people may be different, was originally contributed by NIH research.[143] In fact, research funded by the National Cancer Institute led to a series of discoveries that played important roles in the development of Gleevec.[144] Research as early as the 1950s identified the Philadelphia chromosome, but it was not until the 1970s, when Dr. Nora Heisterkamp, then a researcher at NCI, and her colleagues discovered that when the Philadelphia chromosome forms, two genes that are normally separated get fused together, resulting in the hybrid or fusion gene known as BCR-ABL. Subsequent research by Dr. Owen Witte at UCLA demonstrated that the formation and accumulation of BCR-ABL in the blood stream causes CML.[145]
In the mid-1980s, the Swiss pharmaceutical company Ciba-Geigy initiated a research program directed at the identification of protein kinase inhibitors, a type of regulatory enzyme inhibitor that blocks the action of protein kinases, which turn a protein on or off and therefore affect its level of activity and function.[146] By the early 1990s, Ciba’s more than a half-decade of research into protein kinase inhibitors led its research team to meet Dr. Brian Drucker, then a researcher at the Dana-Farber Cancer Instiute, who was looking to find a cure for CML and believed that BCR-ABL was the key target for addressing the disease.[147] Drucker teamed with Dr. Peter Lydon, who led a drug discovery team at Ciba-Geigy, to further develop and examine applicability of the large inventory of protein kinase inhibitors Ciba-Geigy had been developing. The lead compound that would become imatinib mesylate (Gleevec) was the synthesis of a derivative of 2-phenylamino-pyrimidine that was initially screened against Protein kinase C (a serine-threonine) kinase, not because it was viewed as an ABL kinase inhibitor. However, after some chemical modification that gave the compound the requisite specificity for therapeutic efficiency, the molecular compound CGB-57148B emerged.[148]
Dr. Drucker, who had since moved to the Oregon Health and Science University, began testing the effects of compound CGB-57148B on cancerous cells taken from CML patients in 1993, with the tests demonstrating the compound killed CML cells while leaving normal cells unaffected.[149] Armed with this information, Ciba-Geigy had a decision to make: move forward with compound CGB-57148B, or invest resources in another compound, CGP-53716, which inhibited platelet-derived growth factor receptors (PDGFR) and had the potential to be applicable to a larger cancer patient population.[150] Compound CGB-57148B’s apparent ability to arrest disease progression at the source was appealing, but the perceived small size of the market—Ciba-Geigy analysts estimated a drug based on the compound would produce a mere $100 million in total revenues—inhibited further development of the drug, which was further delayed by Ciba-Geigy’s 1996 merger with Sandoz to form Novartis.[151]
But Daniel Vasella, who became CEO after the merger, was committed to making Novartis an innovation-driven company that creates value by translating breakthrough science into novel new drugs, so he flew in the face of continued cautions from the marketing team and committed Novartis to moving forward with clinical trials in late 1997.[152] Phase I clinical trails of the compound, then identified as STI-571 and later renamed imatinib, which were partly funded by the National Cancer Institutes, began in 1998, and almost immediately began to deliver tremendous results. The drug caused the cancer to disappear in the majority of early-stage patients with CML, with 98 percent of those receiving the drug in trial still in remission after five years.[153] Phase II and III clinical trial work continued during the 2000s, with the FDA approving imatinib in 2011, and Novartis bringing the drug to market as Gleevec.
Today, an individual with CML who has been treated with imatinib and has been in remission for two years has the same life expectancy as someone who does not have cancer. But not only has the drug been a lifesaver for patients with CML, the discovery of imatinib helped establish a group of cancer drugs, called targeted therapies, designed to attack cancer cells with specific genetic abnormalities, which has given rise to the field of precision medicine: treatments tailored to the unique genetic changes in an individual’s cancer cells.[154]
The story of Gleevec illustrates the essential roles played by the public sector, university researchers, and the private sector in bringing a breakthrough, category-defining therapy to market.
The basic scientific research underpinning the Philadelphia chromosome and BCR-ABL’s role as an oncogene stretches back to at least the 1950s, and the story of Gleevec illustrates the essential roles played by the public sector, university researchers, and the private sector. But as Philip Rea, Mark Pauly, and Lawton Burns wrote in their book, Managing Discovery in the Life-sciences: Harnessing Creativity to Drive Biomedical Innovation, the commercialization of Gleevec was no sure thing:
The reluctance of firms to enter the fray even when they had in their hands such a large and path-breaking portfolio for free emphasizes an important point: the danger of proposals to charge profit-maximizing drug firms when they use—pejoratively, “exploit”—the hard won accomplishments of publicly funded research.[155]
In other words, if a government ever had the capacity to march in decades later and compulsorily license the intellectual property underpinning a novel biologic drug on the grounds that some of it may have in part been contributed by federally funded scientific research—and now the government deems the price for that drug “unreasonable”—it would seriously undermine the mechanics of America’s life-sciences innovation system, giving enterprises considerable pause about investing the additional hundreds of millions, even billions, required to bring innovative drugs to the marketplace.
Spark Therapeutics/Luxturna
In 2017, the U.S. Food and Drug Administration approved a treatment for patients suffering from Leber congenital amaurosis (LCA)—a genetic mutation that causes a form of inherited blindness that results in patients suffering severe visual impairment at infancy or early childhood, and often becoming totally blind by midlife—called Luxturna. This was the first gene therapy drug approved by the FDA for the treatment of a genetic disease, and the first drug that injected a new, corrective gene into a patient as part of the treatment.[156] Luxturna also represents the world’s first and only pharmacologic treatment for an inherited retinal disease.
As the National Eye Institute (one of the 27 institutes or centers of the U.S. National Institutes of Health) wrote in its report, “NEI: 50 Years of Advances in Vision Research,” “NEI-supported vision researchers have made great strides in the quest to develop gene therapy treatments for eye disease, particularly for retinal degenerative diseases.”[157] For instance, in the 1990s, Dr. T. Michael Redmond, chief of the NEI Laboratory of Retinal Cell and Molecular Biology, made fundamental discoveries about the molecular biology of the retina, the light-sensing tissue at the back of the eye, including deducing how the RPE65 gene converts dietary vitamin A into a form of the vitamin that is central to the visual cycle, the enzymatic processes by which the eye converts light into electrical signals sent to the brain.[158]
For patients suffering from LCA, the Luxturna gene therapy corrects deficits resulting from mutations of that RPE65 gene, which produces the proteins that make light receptors work in the retina and thus make vision possible. To restore production of those proteins, corrected versions of the RPE65 gene are delivered through a single injection, using a genetically engineered, benign adeno-associated virus to carry the genes directly to the retina.[159]
Work toward developing a gene therapy had begun in the 1990s, with much of the work led by Dr. Jean Bennett and Dr. Albert Maguire, a husband-and-wife team who had conducted over 25 years of research into congenital blindness—including pioneering experiments on mice and canines—alongside other researchers at the Perelman School of Medicine at the University of Pennsylvania and the Children’s Hospital of Philadelphia (CHOP).[160] In the early 2000s, Drs. Bennett and Albert, as well as other NEI-supported researchers, began injecting dogs suffering with a canine form of LCA with a single dose of the RPE65 gene therapy, with the dogs showing significant recovery of vision.[161] By late 2007, a Penn/CHOP-led team began Phase I/II clinical studies with 12 patients, with half experiencing vision improvements within weeks of receiving the therapy. Several children in that initial 2007 study were able to see after only a single shot of gene therapy. At the time, Dr. Bennett hailed the results as remarkable, saying they laid “the foundation for applying gene therapy not only to other forms of childhood-onset retinal disease, but also to more common retinal degenerations.”[162]
Yet gene therapy remained an experimental concept, and the process of bringing a commercial gene therapy solution to market was expensive and daunting. To address this, in 2013, CHOP launched Spark Therapeutics, a separate biotechnology company, to accelerate the development of new gene therapies.[163] Spark took over Phase III clinical trials (which had begun in 2012) and by July 2017 had published four-year Phase III clinical trial data demonstrating the safety and efficacy of Luxturna.[164] In August 2017, final clinical data was published in the journal Clinical and Experimental Ophthalmology confirming the treatment’s efficacy, finding that the therapy dramatically restored most patients’ ability to see, increased their sensitivity to light, and improved their side vision.[165] In December 2017, the FDA approved Luxturna as the first gene therapy for a genetic disease in the United States (and also as an orphan drug). Looking ahead, Spark Therapeutics is focused on discovering, developing, and delivering gene therapies to address a wide range of other genetic diseases, including hemophilia, lysosomal storage disorders, and neurodegenerative diseases.[166] Luxturna provides a case study that shows the importance of federally funded basic research contributing to foundational discoveries that provide a platform for subsequent innovation. It also highlights the subsequent challenge of commercialization and the long timeline involved in completing the clinical trial work of proving the safety and efficacy of new therapies.
The Critical Role of the Bayh-Dole Act
Obtaining the full benefits from federally funded research relies on the effective transfer of knowledge from universities (and national and other federally funded laboratories) to the private sector so that it can be developed into marketable innovations.[167] Here, the 1980 Bayh-Dole Act, which gave universities rights to the intellectual property generated from federal funding, spurred vastly more universities to work more closely with industry, and so created a powerful vehicle for leveraging U.S. investment in basic research into a far stronger engine for commercialization and job creation.[168]
The Bayh-Dole Act spurred many more universities to work more closely with industry, and so created a powerful vehicle for leveraging U.S. investment in basic research into a far stronger engine for commercialization.
Before Bayh-Dole (i.e., pre-1980), the federal government had a very weak track record in commercializing the intellectual property it owned as a result of publicly funded research conducted at universities (or federal laboratories). As late as 1978, the federal government had licensed less than 5 percent of the as many as 30,000 patents it owned.[169] Likewise, throughout the 1960s and 1970s, many American universities shied away from direct involvement in the commercialization of research.[170] Indeed, before the passage of Bayh-Dole, only a handful of U.S. universities even had technology transfer or patent offices.[171] Aware as early as the mid-1960s that the billions of dollars the federal government was investing in R&D was not paying the expected dividends, President Johnson in 1968 asked Elmer Staats, then the comptroller general of the United States, to analyze how many drugs had been developed from NIH-funded research. Johnson was stunned when Staats’s investigation revealed that “not a single drug had been developed when patents were taken from universities [by the federal government].”[172] As his report to Congress elaborated:
At that time we reported that HEW [the Department of Health, Education, and Welfare, predecessor of the Department of Health and Human Services] was taking title for the Government to inventions resulting from research in medicinal chemistry. This was blocking development of these inventions and impeding cooperative efforts between universities and the commercial sector. We found that hundreds of new compounds developed at university laboratories had not been tested and screened by [the] pharmaceutical industry because the manufacturers were unwilling to undertake the expense without some possibility of obtaining exclusive rights to further development of a promising product.[173]
That same year, on Comptroller General Staats’s recommendation, NIH responded by creating the Institutional Patent Agreement (IPA), a program allowing universities with approved technology transfer capabilities to own patents made under agency grants.[174] The effect was immediate. As the Wisconsin Alumni Research Foundation’s Howard Bremer testified before Congress, prior to the IPA program, not one of the University of Wisconsin’s NIH-funded discoveries had ever been licensed. As Bremer stated, “When the government took title, nothing was happening. They went into publications and they went into the literature, and that was the end of it.”[175] After the IPA went into effect, patent applications by universities increased by 300 percent, and of the 329 inventions managed by universities in the first five years, 122 were licensed, including 78 exclusively.[176]
On average, the United States launches approximately three new start-up companies and two new products per day as a result of university inventions brought to market—in part thanks to Bayh-Dole.
The stagflation, economic malaise, and heightened foreign competition America confronted in the 1970s increased the pressure to produce innovation at the highest level, unhindered by burdensome legal code.[177] Policymakers were also aware that the commercialization of discoveries stemming from federally funded research continued to languish. As Joseph P. Allen wrote, “Congress was rightly concerned that potential benefits from billions of dollars of federally funded research were lying dormant on the shelves of government.”[178] Congressmembers were also cognizant that rules about ownership of IP stemming from federal research funding needed to be aligned and streamlined across federal agencies (i.e., not just the IPA’s use by NIH, and briefly by the National Science Foundation).
This need was crystalized when then HEW secretary Joseph Califano suspended the IPA program because he wanted to return to a policy of case-by-case review of agency-funded inventions to determine whether they should be owned by the inventing organization or taken by the government for nonexclusive licensing. Further, in 1975, Califano “marched in” on American Science and Engineering, Inc. (AS&E), a company developing CAT scanners that leveraged two key inventions supported by NIH research funding and which NIH had licensed to AS&E.[179] Califano used march-in provisions to cancel AS&E’s exclusive license, citing concerns that the new CAT scanner technology might increase the costs of health care—even as it improved quality. He also added that by granting invention ownership to universities, the IPA program gave them, not the department, control over the desirability and pace of innovation.[180] Not surprisingly, the pace of subsequent innovation slowed dramatically, as university inventions became entangled in a lengthy bureaucratic process and the development of important treatments was delayed.
This episode was a key catalyst in subsequently leading senators Birch Bayh (D-IN) and Bob Dole (R-KS) to collaborate to introduce legislation allowing universities, small businesses, and nonprofit institutions to take ownership of intellectual property rights stemming from discoveries made with federally funded research. As Joseph P. Allen noted, HEW’s “attempt to march in to control product prices had an ironic result: it undermined the presumption that Washington knew best how to license federally funded inventions.”[181] Or, as John Rabitschek and Norman Latker noted, as Bayh-Dole was being debated in Congress, “A broad political consensus ultimately developed around the notion that market forces would do a better job of commercializing government-funded technology than federal agencies could.”[182] By enacting Bayh-Dole, Congress decentralized patent management from the federal bureaucracy into the hands of the inventing organizations. As the first words of the Bayh-Dole Act explicitly noted, “It is the policy and objective of the Congress to use the patent system to promote the utilization of inventions arising from federally supported research and development.”[183]
Again, the impact was immediate. The Bayh-Dole Act has been widely praised as a significant factor contributing to the United States’ “competitive revival” in the 1990s.[184] In 2002, The Economist called Bayh-Dole:
Possibly the most inspired piece of legislation to be enacted in America over the past half-century. Together with amendments in 1984 and augmentation in 1986, this unlocked all the inventions and discoveries that had been made in laboratories throughout the United States with the help of taxpayers’ money. More than anything, this single policy measure helped to reverse America's precipitous slide into industrial irrelevance.[185]
Allowing U.S. institutions to earn royalties through the licensing of their research provided a powerful incentive for universities and other institutions to pursue commercialization opportunities.[186] The Bayh-Dole Act almost immediately led to an increase in academic patenting activity. For instance, while only 55 U.S. universities had been granted a patent in 1976, 240 universities had been issued at least one patent by 2006.[187] Similarly, while only 390 patents were awarded to universities in 1980, by 2009, that number had increased to 3,088—and by 2015, to 6,680. Another analysis found that in the first two decades of Bayh-Dole (i.e., 1980 to 2002) American universities experienced a tenfold increase in their patents, and created more than 2,200 companies to exploit their technology.[188] In total, over 80,000 U.S. patents have been issued to academic research institutions over the past 25 years.[189] Moreover, academic technology transfer has supported the launch of over 12,000 start-ups since 1995.[190] According to a report prepared for AUTM and the Biotechnology Industry Organization (BIO), from 1996 to 2015, academic patents and their subsequent licensing to industry—substantially stimulated by the Bayh-Dole Act—bolstered U.S. GDP by up to $591 billion, contributed to $1.3 trillion in gross U.S. industrial output, and supported 4,272,000 person years of employment.[191] More than 200 drugs and vaccines have been developed through public-private partnerships since the Bayh-Dole Act entered force in 1980.[192]
AUTM’s 2016 Annual Survey again reiterated what a significant force academic technology transfer is in driving economic development. It found that the number of invention disclosures increased to 25,825 over the previous five years. From these discoveries, 16,487 new U.S. patent applications were filed, leading to 7,021 U.S. patents issued. It also identified 1,024 new start-ups formed on the basis of technology transfer.[193] In fact, on average, three new start-up companies and two new products are launched in the United States every day as a result of university inventions brought to market, in part thanks to the Bayh-Dole Act.[194] As Harvard University’s Naomi Hausman wrote, “The sort of large scale technology transfer from universities that exists today would have been very difficult and likely impossible to achieve without the strengthened property rights, standardized across granting agencies, that were set into law in 1980.”[195]
The Bayh-Dole Act has produced a number of additional benefits. For example, Hausman analyzed the impact of Bayh-Dole in shaping university relations with local economies, and found that the increase in university connectedness to industry under the intellectual property regime created by Bayh-Dole produced important local economic benefits. In particular, Hausman found that long-run employment, payroll, payroll per worker, and average establishment size grew differentially more after the 1980 Bayh-Dole Act in industries more closely related to innovations produced by a local university or hospital.[196] There is also evidence that the Bayh-Dole Act contributed to university faculty responding to royalty incentives by producing higher-quality innovations.[197] Evidence further suggests that patenting increased most after Bayh-Dole in lines of business that most value technology transfer via patenting and licensing.[198]
Many countries have enacted Bayh-Dole-like legislation because they recognize its power in turning their universities into engines of innovation.
Finally, countries throughout the world—including Brazil, China, Indonesia, Japan, Korea, Malaysia, the Philippines, Singapore, South Africa, and Taiwan—have since followed the United States’ lead in establishing policies that grant their universities IP ownership rights.[199] Even Kazakhstan and Zimbabwe are currently looking at implementing Bayh-Dole-like legislation, recognizing its power to help turn their universities into engines of innovation and commercialization. Likewise, the California Senate Office of Research conducted a comprehensive analysis of the Bayh-Dole Act and concluded: “After reviewing the literature and interviewing key experts, we recommend the Legislature consider adopting a statewide IP policy replicating the principles of the Bayh–Dole Act for research granting programs.”[200] U.S. states and foreign countries have supported adoption of Bayh-Dole-like policies because they recognize that Bayh-Dole works. Simply put, the Bayh-Dole Act has created a powerful engine of practical innovation, producing many scientific advances that have extended human life, improved its quality, and reduced suffering for millions of people.[201]
The Risks Inappropriate Use of March-In Rights Pose to the Bayh-Dole Act
In short, the Bayh-Dole Act has been an unparalleled success. Yet some have advocated for policies that would undermine some of its key provisions and effects. At issue are the so-called march-in rights, a provision within the Bayh-Dole Act that permits the government, in specified circumstances, to require patent holders to grant a “nonexclusive, partially exclusive, or exclusive license” to a “responsible applicant or applicants.”[202] As the following section explains, the architects of the Bayh-Dole Act principally intended for march-in rights to be used to ensure patent owners commercialized their inventions.[203] As Senator Birch Bayh explained:
When Congress was debating our approach fear was expressed that some companies might want to license university technologies to suppress them because they could threaten existing products. Largely to address this fear, we included the march-in provisions.[204]
Yet a number of civil society organizations and some members of Congress have called on the National Institutes of Health to exploit Bayh-Dole march-in rights to “control” allegedly unreasonably high drug prices. Six petitions requesting NIH to “march in” with respect to a particular pharmaceutical drug have been filed.[205] In four of these cases, the petition was filed by civil society organizations alleging that a company was pricing a drug too high.[206] Some 50 members of Congress, led by Representative Lloyd Doggett (D-TX), have called on NIH to cancel exclusivity when patented drugs are not available with reasonable terms.[207] Senator Angus King (I-ME) proposed legislation in 2017 that would require the Department of Defense (DOD) to issue compulsory licenses under Bayh-Dole “whenever the price of a drug, vaccine, or other medical technology is higher in the U.S. than the median price charged in the seven largest economies that have a per capita income at least half the per capita income of the U.S.”[208] In other words, DOD would force a licensor to divulge their intellectual property so that a drug could be manufactured by other licensees, and in theory be sold at a lower price. (While it was not enacted, a similar provision was unfortunately included in the FY 2018 National Defense Authorization Act (NDAA) Senate Armed Services Committee report.) Yet the Bayh-Dole Act’s designers did not intend for march-in rights to be used to control drug prices.
Bayh-Dole March-In Rights Were Never Intended to Address Drug Price Concerns
The Bayh-Dole Act proscribes four specific instances in which the government is permitted to exercise march-in rights:
1. If the contractor or assignee has not taken, or is not expected to take within a reasonable time, effective steps to achieve practical application of the subject invention;
2. If action is necessary to alleviate health or safety needs not reasonably satisfied by the patent holder or its licensees;
3. If action is necessary to meet requirements for public use specified by federal regulations and such requirements are not reasonably satisfied by the contractor, assignee, or licensees; or
4. If action is necessary, in exigent cases, because the patented product cannot be manufactured substantially in the United States.[209]
In other words, lower prices are not one of the rationales laid out in the act. In fact, as senators Bayh and Dole have themselves noted, the Bayh-Dole Act’s march-in rights were never intended to control or ensure “reasonable prices.”[210] As the twain wrote in a 2002 Washington Post op-ed titled, “Our Law Helps Patients Get New Drugs Sooner,” the Bayh-Dole Act:
Did not intend that government set prices on resulting products. The law makes no reference to a reasonable price that should be dictated by the government. This omission was intentional; the primary purpose of the act was to entice the private sector to seek public-private research collaboration rather than focusing on its own proprietary research.[211]
The op-ed reiterated that the price of a product or service was not a legitimate basis for the government to use march-in rights, noting:
The ability of the government to revoke a license granted under the act is not contingent on the pricing of a resulting product or tied to the profitability of a company that has commercialized a product that results in part from government-funded research. The law instructs the government to revoke such licenses only when the private industry collaborator has not successfully commercialized the invention as a product.[212]
Rather, Bayh-Dole’s march-in provision was designed as a fail-safe for limited instances in which a licensee might not be making good-faith efforts to bring an invention to market, or when national emergencies require that more product is needed than a licensee is capable of producing. As Joseph P. Allen, a senate staffer for Bayh who played a key role in shaping the legislation, explains, Congress’s introduction of Bayh-Dole was intended “to decentralize patent management from the bureaucracy into the hands of the inventing organizations, while retaining the long-established precedent that march-in rights were to be used in rare situations when effective efforts are not being made to bring an invention to the marketplace or enough of the product is not being produced to meet public needs.”[213]
Likewise, the National Institute of Standards and Technology (NIST) report “Return on Investment Initiative: Draft Green Paper” agrees, noting, “The use of march-in is typically regarded as a last resort, and has never been exercised since the passage of the Bayh-Dole Act in 1980.”[214] The report notes that, “NIH determined that that use of march-in to control drug prices was not within the scope and intent of the authority.”[215]
March-in rights have never been exercised during the 39-year history of the Bayh-Dole Act
In fact, there has only been one case in which Bayh-Dole’s march-in criteria truly would have been met: a 2010 case in which Genzyme encountered difficulties in manufacturing sufficient quantities of Fabrazyme/agalsidase beta, an orphan drug for the treatment of Fabry disease.[216] Genzyme had to shut down the plant making the drug due to quality control issues and was therefore unable to manufacture the drug in sufficient quantities. NIH investigated the situation but did not initiate a march-in proceeding because it found that “Genzyme was working diligently to resolve its manufacturing difficulties” and that the company was likely to get back into production faster than a new licensee could get FDA approval to make the drug.[217]
March-in rights have never been exercised during the 39-year history of the Bayh-Dole Act.[218] NIH has denied all six petitions to apply march-in rights, noting that the drugs in question were in virtually all cases adequately supplied and that concerns over drug pricing were not, by themselves, sufficient to provoke march-in rights.[219] NIH itself has expressed skepticism about the use of march-in rights to control drug prices, noting:
Finally, the issue of the cost or pricing of drugs that include inventive technologies made using federal funds is one which has attracted the attention of Congress in several contexts that are much broader than the one at hand. In addition, because the market dynamics for all products developed pursuant to licensing rights under the Bayh-Dole Act could be altered if prices on such products were directed in any way by NIH, the NIH agrees with the public testimony that suggested that the extraordinary remedy of march-in is not an appropriate means of controlling prices.[220]
As Rabitschek and Latker wrote in “Reasonable Pricing—A New Twist for March-in Rights Under the Bayh-Dole Act” in the Santa Clara University High Technology Law Journal, “A review of the [Bayh-Dole] statute makes it clear that the price charged by a licensee for a patented product has no direct relevance to march-in rights.”[221] As the authors concluded:
There is no reasonable pricing requirement under 35 U.S.C. §203(l)(a)(1), considering the language of this section, the legislative history, and the prior history and practice of march-in rights. Rather, this provision is to assure that the contractor utilizes or commercializes the funded invention.[222]
The argument that Bayh-Dole march-in rights could be used to control drug prices was originally advanced in an article by Peter S. Arno and Michael H. Davis.[223] They contended that “[t]he requirement for ‘practical application’ seems clear to authorize the federal government to review the prices of drugs developed with public funding under Bayh-Dole terms and to mandate march-in when prices exceed a reasonable level” and suggested that under Bayh-Dole, the contractor may have the burden of showing that it charged a reasonable price.[224] While Arno and Davis admitted there was no clear legislative history on the meaning of the phrase “available to the public on reasonable terms,” they still concluded that, “[t]here was never any doubt that this meant the control of profits, prices, and competitive conditions.”[225]
But as Rabitschek and Latker explain, there are several problems with this analysis. First, the notion that “reasonable terms” of licensing means “reasonable prices” arose in unrelated testimony during the Bayh-Dole hearings. Most importantly, they note, “If Congress meant to add a reasonable pricing requirement, it would have explicitly set one forth in the law, or at least described it in the accompanying reports.”[226] As Rabitschek and Latker continue, “There was no discussion of the shift from the ‘practical application’ language in the Presidential Memoranda and benefits being reasonably available to the public, to benefits being available on reasonable terms under 35 U.S.C. § 203.”[227] As they conclude, “The interpretation taken by Arno and Davis is inconsistent with the intent of Bayh-Dole, especially since the Act was intended to promote the utilization of federally funded inventions and to minimize the costs of administering the technology transfer policies…. [The Bayh-Dole Act] neither provides for, nor mentions, ‘unreasonable prices.’”[228]
Again, the Bayh-Dole Act’s march-in provisions were included with commercialization in mind. Related to this, another reason the Bayh-Dole Act’s architects inserted march-in right provisions was because, at the time the law was introduced, very few universities were experienced in patent licensing. The march-in provision therefore served as a fail-safe in cases in which universities were not effectively monitoring their agreements.[229] But universities have in fact proven proactive and effective in enforcing their licensing agreements, regularly including development milestones in their licenses—and when these milestones aren’t being met without satisfactory reason (e.g., development is more difficult than expected), universities often terminate the deal and look for another developer. In other words, universities are enforcing their licensing agreements, not letting licenses just sit on the technologies—another example of why there has been no reason for the government to march in.
Allowing March-In Rights to Address Pricing Would Lead to Significantly Reduced Innovation
Even if Congress were to amend Bayh-Dole to allow the federal government to use march-in rights to force lower pricing, the result would be a reduction of life-sciences innovation, as past experience history clearly shows.
The debate around “reasonable pricing” of drugs stemming from licensed research goes back some time. The Federal Technology Transfer Act of 1986 (FTTA) authorized federal laboratories to enter into Cooperative Research and Development Agreements (CRADAs) with numerous entities, including private businesses. NIH has found that CRADAs “significantly advance biomedical research by allowing the exchange and use of experimental compounds, proprietary research materials, reagents, scientific advice, and private financial resources between government and industry scientists.”[230]
Proposals for forced requirements of reasonable pricing in life-sciences IP licensing have consistently led to less—not more—innovation.
In 1989, NIH’s Patent Policy Board adopted a policy statement and three model provisions to address the pricing of products licensed by public health service (PHS) research agencies on an exclusive basis to industry, or jointly developed with industry through CRADAs. In doing so, the Department of Health and Human Services (DHHS) became the only federal agency at the time (other than the Bureau of Mines) to include a “reasonable pricing” clause in its CRADAs and exclusive licenses.[231] The 1989 PHS CRADA Policy Statement asserted:
DHHS has a concern that there be a reasonable relationship between pricing of a licensed product, the public investment in that product, and the health and safety needs of the public. Accordingly, exclusive commercialization licenses granted for the NIH/ADAMHA [Alcohol, Drug Abuse, and Mental Health Administration] intellectual property rights may require that this relationship be supported by reasonable evidence.
But as Joseph P. Allen noted, such “attempts to impose artificial ‘reasonable pricing’ requirements on developers of government supported inventions did not result in cheaper drugs. Rather, companies simply walked away from partnerships.”[232] Use of CRADAs began in 1987 and rapidly increased until the reasonable pricing requirement hit in 1989, after which they declined through 1995 (see figure 4).
Figure 5: Private-Sector CRADAs With NIH, 1987–2017[233]
Recognizing that the only impact of the reasonable pricing requirement was undermining scientific cooperation without generating any public benefits, NIH eliminated the reasonable pricing requirement in 1995. In removing the requirement, then NIH director Dr. Harold Varmus explained, “An extensive review of this matter over the past year indicated that the pricing clause has driven industry away from potentially beneficial scientific collaborations with PHS scientists without providing an offsetting benefit to the public. Eliminating the clause will promote research that can enhance the health of the American people.”[234] As figure 4 shows, after NIH eliminated the requirement in 1995, the number of CRADAs immediately rebounded in 1996, and grew considerably in the following years.[235] The case represents a natural experiment showing the harm pricing requirements can inflict. Somewhat similarly, as the California Senate Office of Research has noted, “Granting agencies such as the National Institutes of Health (NIH) ultimately have abandoned policies that require a financial return to the government after concluding that removing barriers to the rapid commercialization of products represents a greater public benefit than any potential revenue stream to the government.”[236]
A more recent case involved biopharmaceutical company Sanofi possibly taking a license from the U.S. Army to develop a vaccine for the Zika virus. U.S. Army scientists from the Walter Reed Army Institute of Research developed two candidate vaccines for the Zika virus and posted a notice in the federal register offering to license them on either a nonexclusive or exclusive basis. No company responded to the nonexclusive license, and Sanofi was the only company that submitted a license application for the Army’s Zika candidate vaccine, with the U.S. Army and Sanofi reaching a licensing agreement in June 2016 that would enable Sanofi to continue the development and clinical trial work necessary to turn the candidate vaccine into a market-ready product. As a U.S. Army official noted, “Exclusive licenses are often the only way to attract a competent pharma partner for such development projects,” and are needed because the military lacks “sufficient” research and production capabilities to develop and manufacture a Zika vaccine.[237] Sanofi received a $43 million government grant to start undertaking clinical trial work on the virus candidate.
In July 2017, supported by Knowledge Economy International, an organization opposed to intellectual property rights, Sens. Bernie Sanders (D-VT) and Rick Durbin (D-IL) argued that the U.S. Army and Sanofi should insert reasonable pricing language into the exclusive license. Sanders even called on President Trump to cancel the deal.[238] In response, Army officials noted that they were not in a position to “enforce future vaccine prices.” For its part, Sanofi representatives noted, “We can’t determine the price of a vaccine that we haven’t even made yet,” and argued that “it’s premature to consider or predict Zika vaccine pricing at this early stage of development. As noted earlier, ongoing uncertainty around epidemiology and disease trajectory make any commercial projections theoretical at best.”[239] Sanofi noted that it had committed over 60 researchers to the effort, invested millions of dollars itself, and was “committed to leveraging its flavivirus vaccine development and manufacturing expertise to deliver and ultimately price a Zika vaccine in a responsible way.”[240]
Sanofi also noted that the proposed license would require it to pay milestone and royalty payments back to the government, and its exclusive license would not prevent other companies—such as GlaxoSmithKline, Takeda, and Moderna, which also had all struck their own Zika vaccine partnerships with U.S. agencies—from bringing competing products to market, and allow for robust competition in the market for Zika vaccines.[241] However, with both partners continuing to be attacked in the media, in September 2017, Sanofi announced it would “not continue development of, or seek a license from, the Walter Reed Army Institute of Research for the Zika vaccine candidate at this time.”[242] This is yet another case wherein certain policymakers’ insistence on pricing requirements stifled innovation and the potential for a firm to bring a promising innovation to market.
There are over 200 new drugs and vaccines on the worldwide market today as a result of Bayh-Dole, compared with zero before—a fact opponents fail to acknowledge.
It also takes the debate back to the central point that, in their push for lower drug prices through weaker private IP rights stemming from federally funded research, advocates fail to realize that no drugs were created from federally funded inventions under the previous (to Bayh-Dole) regime.[243] In contrast, over 200 new drugs and vaccines have been developed through public-private partnerships facilitated in part by the Bayh-Dole Act since its enactment in 1980.[244]
Weakening the certainty of access to IP rights provided under Bayh-Dole by employing march-in to address drug pricing issues—especially if it meant a government entity could walk in and retroactively commandeer innovations private-sector enterprises invested hundreds of millions, if not billions, to create—would significantly diminish private businesses’ incentives to commercialize products supported by federally funded research.[245] As David Bloch notes, “The reluctance of such [biopharmaceutical] companies to do business with the government is almost invariably tied up in concerns over the government’s right to appropriate private sector intellectual property.”[246] As he continues, “Each march-in petition potentially puts at risk the staggeringly massive investment that branded pharmaceutical companies make in developing new drug therapies.”[247] Put simply, using march-in rights to control drug prices could seriously compromise medical discovery and innovation.
For this reason policymakers should resist calls to misuse Bayh-Dole march-in provisions or to create new provisions to limit IP rights to “control” drug prices. For example, NIST’s Green Book on federal technology transfer observed, “RFI respondents had generally positive views of the NIH’s march-in determinations and thought NIH’s approach appropriate.”[248] In reviewing how Bayh-Dole could be improved, the Green Book stated the federal government would, “implement regulatory change under the Bayh-Dole Act by specifying that march-in rights should not be used as a mechanism to control or regulate the market price of goods and services.”[249] The Green Book also avowed to clarify the statute’s meaning of “reasonable terms” and “practical application” in ways that would allow flexibility in crafting commercial or other terms in license agreements to achieve effective technology transfer.
NIST’s Green Book also addresses the government use license, which refers to the “nonexclusive, nontransferable, irrevocable, paid-up license to practice the invention or have the invention practiced throughout the world by or on behalf of the Government,” that applies to any federally funded invention.[250] It observed that respondents “noted that an overly broad interpretation of this right was contrary to the stated intent of the Bayh-Dole Act to allow rights to be elected and retained by the contractor.” The report also addressed application of the government use license to obtain discounts on products developed from federally funded R&D, primarily pharmaceuticals, citing a 2003 Government Accountability Office (GAO) report which concluded that the government use license does not bestow the broader right to purchase royalty-free (i.e., discounted) products that happen to incorporate a federally funded invention if not produced under the government’s license.[251] NIST’s Green Book stated an intended action to “define the scope of the “government use license” for use directly by the government—or a government contractor in the performance of an agreement with the government—for a government purpose only, including continued use in research and development by the government.”[252]
Finally, while adding reasonable price terms to march-in rights legislation could in theory reduce prices by spurring more competition, the reality is it would not do that because companies would not engage with universities in licensing technologies in the first place if they were aware that the risk to their investments would be extremely high. This is exactly what we have seen over the last 60 years when such rights were not in place or were threatened to be taken away.
Advocates for lower drug prices would say that is fine: If the original company does not want to license the technology, then just let any and all comers license it. However, the reality is universities would license less because companies would be reluctant to take non-exclusive licenses, and so universities would therefore lose license fees and ultimately receive less money to support research, develop research labs, train students, etc.
The Bayh-Dole Act and U.S. International Life-Sciences Competitiveness
As this report noted in addressing the history and impact of the Bayh-Dole Act, it’s important to reemphasize that the Bayh-Dole Act was introduced, in part, as a response to deteriorating U.S. economic competitiveness in the late 1970s and early 1980s in the face of heightened foreign competition. And while this report has placed the Bayh-Dole Act as one of a suite of policies that have turned the United States into the world’s leader in life-sciences innovation, it’s important to recognize that foreign competition remains intense and that U.S. leadership is not assured, especially if it were to start implementing policies that could undermine America’s leadership position (such as applying Bayh-Dole march-in rights for the purposes of controlling drug prices).
Indeed, a number of economic competitors, including Australia, China, Japan, Singapore, and the United Kingdom, have recently introduced concerted national strategies to enhance their life-sciences competitiveness. In a recent review of efforts by several countries, TEConomy Partners found that, “Although the United States continues to rank first in nearly all measures of innovation, the countries profiled continue to make significant efforts to try to close the gap with the United States.”[253] That report continued, “The trends over the past five years continue to suggest that, in all but a few areas, the United States is not keeping pace and is actually losing ground.”[254] For instance, according to the OECD, of 25 nations, the United States ranked second from last in inflation-adjusted growth in health research expenditures from 2006 to 2015. Only Italy (-27 percent) was below the United States (-1 percent). In contrast, over that time period, Denmark increased its health research funding by 185 percent, South Korea by 177 percent, Germany by 65 percent, the United Kingdom by 55 percent, and Spain by 32 percent.[255]
Life-sciences competition from China, in particular, is increasingly fierce. In its “Made in China 2025” plan, the Chinese government identified the life-sciences industry as one of 10 key strategic industries it wants to advance, and the country is investing billions toward that cause. One key way Chinese governments are supporting the industry is by subsidizing research parks and providing venture capital. Indeed, a large number of biomedical industry research parks have recently sprung up, and there were already 111 biological science research parks in China as of 2018. For example, Shanghai’s “Pharma Valley”—a 10-square-kilometer life-sciences hub—is now home to more than 500 biotech companies. Further, China’s Ministry of Science and Technology has committed to building 10 to 20 biomedicine science parks by 2020 with a total output value surpassing 10 billion yuan ($1.45 billion).[256] The Chinese government is also investing significant amounts of money in key research areas. For instance, in 2010, the China Development Bank provided a $1.58 billion line of credit to the Beijing Genomics Institute (BGI), a private genome sequencing center, to buy 128 advanced DNA sequencing machines. The investment made BGI the world’s largest genetic sequencer, accounting for roughly one-quarter of all DNA data sequenced in the world in 2014.[257] Further, in 2017, China devoted $9.2 billion to a 15-year Precision Medicine Initiative that seeks to map 100 million human genomes.
The point here is that global life-sciences competition only continues to intensify, and U.S. leadership going forward is no sure thing, especially if it fails to continue supporting the types of policies that have contributed immensely to that leadership. The Bayh-Dole Act represents one of these core policies, and its effectiveness today is as important as it ever has been in underpinning U.S. life-sciences leadership.
Conclusion
The United States leads the world in life-sciences innovation because, over the past four decades, it has implemented a comprehensive set of policies designed to make America the most attractive location in the world to conduct life-sciences R&D. The 1980 Bayh-Dole Act—in giving universities, small businesses, and other nonprofit research institutions rights to the intellectual property stemming from federally funded R&D—has proven to be a critical enabler of this system, and has significantly amplified the utilization of federally funded research. Before Bayh-Dole, the vast majority of discoveries made from federally funded research sat on the shelf and were simply not commercialized or brought to market in the form of products that changed lives—a situation that was especially true of the life-sciences sector.
The Bayh-Dole Act is working as its architects intended, and does not require changing. (Mis)using march-in rights or establishing new ones in order to control drug prices would result in fewer new drugs. Companies would be highly reticent to spend billions developing a drug if they knew the government could come in as long as two decades later and seize or compulsorily license their IP in order to control drug prices. As this report has shown, proposals for forced requirements of reasonable pricing in life-sciences IP licensing have consistently led to less—not more—innovation. And rather than leading to lower prices, such requirements would cause the pipeline of technology transfer to dry up. The resulting reduction in drugs coming to market would impede market-based competition, which also plays a role in managing costs. In conclusion, policymakers should reject proposals that would enable the government to use Bayh-Dole Act march-in provisions for the purpose of controlling drug prices.
Acknowledgments
The author wishes to thank Dr. Robert Atkinson, Val Giddings, and Nigel Cory for providing input to this report. Any errors or omissions are the author’s alone.
About The Author
Stephen J. Ezell is ITIF vice president for Global Innovation Policy and focuses on science, technology, and innovation policy as well as international competitiveness and trade policy issues. He is the coauthor of Innovating in a Service Driven Economy: Insights Application, and Practice (Palgrave McMillan, 2015) and Innovation Economics: The Race for Global Advantage (Yale 2012).
About ITIF
The Information Technology and Innovation Foundation (ITIF) is an independent 501(c)(3) nonprofit, nonpartisan research and educational institute that has been recognized repeatedly as the world’s leading think tank for science and technology policy. Its mission is to formulate, evaluate, and promote policy solutions that accelerate innovation and boost productivity to spur growth, opportunity, and progress. For more information, visit itif.org/about.
Endnotes
[1]. Ross C. DeVol, Armen Bedroussian, and Benjamin Yeo, “The Global Biomedical Industry: Preserving U.S. Leadership” (Milken Institute, September 2011, 5), https://www.lifechanginginnovation.org/sites/default/files/files/Global%20Bio_Full%20Report_WEB.pdf.
[2]. Joe Kennedy, “How to Ensure That America’s Life-Sciences Sector Remains Globally Competitive” (Information Technology and Innovation Foundation, March 2018), 15, https://itif.org/publications/2018/03/26/how-ensure-americas-life-sciences-sector-remains-globally-competitive; European Federation of Pharmaceutical Industries and Associations (EFPIA), “The Pharmaceutical Industry in Figures, Key Data 2017” (EFPIA, 2017), 8, https://www.efpia.eu/media/219735/efpia-pharmafigures2017_statisticbroch_v04-final.pdf.
[3]. David Michels and Aimison Jonnard, “Review of Global Competitiveness in the Pharmaceutical Industry” (U.S. International Trade Commission, 1999), 2-3, https://www.usitc.gov/publications/332/pub3172.pdf.
[4]. John K. Jenkins, M.D., “CDER New Drug Review: 2015 Update” (PowerPoint presentation, U.S. Food and Drug Administration/CMS Summit, Washington, D.C., December 14, 2015), http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM477020.pdf.
[5]. European Federation of Pharmaceutical Industries and Associations, “The Research Based Pharmaceutical Industry: A Key Actor for a Healthy Europe” (EFPIA, 2006).
[6]. Shanker Singham, “Improving U.S. Competitiveness—Eliminating Anti-Market Distortions” (working paper, International Roundtable on Trade and Competition Policy, 2012).
[7]. Jenkins, “CDER New Drug Review,” 23.
[8]. Ibid.
[9]. Justin Chakma et al., “Asia’s Ascent—Global Trends in Biomedical R&D Expenditures,” New England Journal of Medicine 370, No. 1 (January 2014); ER Dorsey et al., “Funding of US Biomedical Research, 2003-2008” New England Journal of Medicine 303 (2010): 137–43, http://www.ncbi.nlm.nih.gov/pubmed/20068207.
[10]. Pharmaceutical Research and Manufacturers of America (PhRMA), “Executive Summary: The Biopharmaceutical Pipeline: Innovative Therapies in Clinical Development” (PhRMA, 2017), 1, http://phrma-docs.phrma.org/files/dmfile/PhRMA_Biopharma_ExecutiveSummary_FINAL.pdf.
[11]. Stephen Ezell, “How the Prescription Drug User Fee Act Supports Life-Sciences Innovation and Speeds Cures” (Information Technology and Innovation Foundation, February 2017), http://www2.itif.org/2017-pdufa-life-sciences-innovation.pdf; United States General Accountability Office (GAO), “FDA Drug Approval: Review Time Has Decreased in Recent Years” (Washington, D.C.: GAO, October 20, 1995), http://www.gao.gov/assets/230/221919.pdf; Jenkins, “CDER New Drug Review.”
[12]. Stephen Ezell, “Comments to the Centers for Medicare & Medicaid Services on an International Pricing Index Model for Medicare Part B Drugs” (Information Technology and Innovation Foundation, December 2018), http://www2.itif.org/2018-CMS-pricing-index-comments.pdf.
[13]. Organization for Economic Cooperation and Development (OECD), Pharmaceutical Pricing Policies in a Global Market (OECD, September 2008), 190, http://www.oecd.org/els/pharmaceutical-pricing-policies-in-a-global-market.htm.
[14]. Robert D. Atkinson, “Why Life-Sciences Innovation Is Politically ‘Purple’—and How Partisans Get It Wrong” (Information Technology and Innovation Foundation, February 2016), http://www2.itif.org/2016-life-sciences-purple.pdf.
[15]. Dr. Everett Ehrlich, “An Economic Engine: NIH Research, Employment, and the Future of the Medical Innovation Sector” (United for Medical Research, 2011), 3, http://www.eyeresearch.org/pdf/UMR_Economic%20Engine_042711a.pdf.
[16]. National Institutes of Health, “Institutes, Centers, and Offices,” https://www.nih.gov/institutes-nih.
[17]. Judith A. Johnson and Kavya Sekar, “NIH Funding: FY1994–FY2019” (Congressional Research Service, October 2018), 1, https://fas.org/sgp/crs/misc/R43341.pdf; Jocelyn Kaiser, “NIH Gets $2 Billion Boost in Final 2019 Spending Bill,” Science, September 14, 2018, https://www.sciencemag.org/news/2018/09/nih-gets-2-billion-boost-final-2019-spending-bill.
[18]. Johnson and Sekar, “NIH Funding: FY1994-FY2019,” 1.
[19]. Pierre Azoulay et al., “Public R&D Investments and Private-sector Patenting: Evidence from NIH Funding Rules,” NBER Working Paper 16–056 (January 2017), https://www.nber.org/papers/w20889.
[20]. U.S. National Institutes of Health, “Turning Discovery Into Health: Impact On Our Nation,” (NIH, December 2017), https://www.nih.gov/sites/default/files/about-nih/impact/impact-our-nation.pdf.
[21]. Ibid.
[22]. Ehrlich, “An Economic Engine,” 3.
[23]. Federation of American Societies for Experimental Biology (FASEB), “Federal Funding for Biomedical and Related Life-sciences Research” (FASEB, 2017), https://www.faseb.org/Portals/2/PDFs/opa/2016/Federal_Funding_Report_FY2017_FullReport.pdf.
[24]. Ruchika Nijhara et al., “Bypassing Bypass Surgery and Other Success Stories from the National Institutes of Health,” Journal of the Association of University Technology Managers XVII, No. 2 (2005), http://www.ott.nih.gov/pdfs/Ferguson-AUTM-Journal-vol-17-no-2-pp-1-16-Fall-2005.pdf.
[25]. Carolyn Treasure, “Do March-in Rights Ensure Low-Cost Access to Medical Products Arising From Federally-Funded Research? A Qualitative Study” (doctoral dissertation, Harvard Medical School, 2016), 5, https://dash.harvard.edu/handle/1/27007723.
[26]. Foundation for the National Institutes of Health (FNIH), “FNIH Capabilities Brochure,” (FNIH, 2018), 1, https://fnih.org/content/fnih-capabilities-brochure.
[27]. Foundation for the National Institutes of Health, “FNIH 2017 Annual Report,” (FNIH, 2017), https://fnih.org/2017-annual-report/about-us/.
[28]. Foundation for the National Institutes of Health, “Biomarkers Consortium,” https://fnih.org/what-we-do/biomarkers-consortium.
[29]. PhRMA, “Chart Pack: Biopharmaceuticals in Perspective: Summer 2018,” (PhRMA, 2018), 35, https://www.phrma.org/report/chart-pack-biopharmaceuticals-in-perspective-summer-2018.
[30]. National Institutes of Health, National Center for Advancing Translational Sciences, “Center Overview,” https://ncats.nih.gov/about/overview.
[31]. National Institutes of Health, National Center for Advancing Translational Sciences, “About the Center,” https://ncats.nih.gov/about/center.
[32]. National Institutes of Health, National Center for Advancing Translational Sciences, “Transforming Translational Science,” https://ncats.nih.gov/files/NCATS-factsheet.pdf.
[33]. National Institutes of Health, “What Are SBIR and STTR Programs?” https://sbir.nih.gov/.
[34]. Matthew Portnoy, “HHS SBIR/STTR PHS 2017-2 Grant Omnibus Webinar” (PowerPoint presentation, June 29, 2017), https://sbir.nih.gov/sites/default/files/FY17%20HHS%20SBIR-STTR%20Grant%20Omnibus%20Webinar%20-%20Slide%20Presentation%20-%206.29.17.pdf.
[35]. Jere Glover, “Testimony Before the Committee on Small Business and Entrepreneurship, United States Senate, Reauthorization of the SBIR/STTR Programs—The Importance of Small Business Innovation to National and Economic Security,” January 28, 2016.
[36]. National Academies Press, “SBIR/STTR at the National Institutes of Health,” https://www.ncbi.nlm.nih.gov/books/NBK338147/.
[37]. National Institutes of Health, National Heart Lung, and Blood Institute, “About NCAI and REACH,” https://ncai.nhlbi.nih.gov/ncai/aboutncai/mission.
[38]. The Association of the British Pharmaceutical Industry (ABPI), “Worldwide Pharmaceutical Company R&D Expenditure,” accessed January 21, 2019, https://www.abpi.org.uk/facts-and-figures/science-and-innovation/worldwide-pharmaceutical-company-rd-expenditure/.
[39]. International Federation of Pharmaceutical Manufacturers & Associations (IFPMA), The Pharmaceutical Industry and Global Health: Facts and Figures 2014 (IFPMA, 2014), 13, http://www.ifpma.org/fileadmin/content/Publication/2014/IFPMA_-_Facts_And_Figures_2014.pdf; PhRMA, “Economic Impact,” http://www.phrma.org/economic-impact.
[40]. Joe Kennedy, “How to Ensure That America’s Life-Sciences Sector Remains Globally Competitive”; National Science Foundation, “Scientific and Engineering Indicators 2018,” (NSF, 2018): 4–73, https://www.nsf.gov/statistics/2018/nsb20181/assets/nsb20181.pdf.
[41]. PhRMA, “Chart Pack: Biopharmaceuticals in Perspective,” Version 2.0, (Washington, D.C.: PhRMA, Spring 2012).
[42]. ABPI, “Worldwide Pharmaceutical Company R&D Expenditure.”
[43]. Pacific BioLabs, “Stages of Drug Development,” https://pacificbiolabs.com/stages-of-drug-development.
[44]. PhRMA, “Chart Pack: Biopharmaceuticals in Perspective: Summer 2018,” 29.
[45]. Ibid.
[46]. The lower numbers are from a study by the U.K. Office of Health Economics: Jorge Mestre-Ferrandiz, Jon Sussex, and Adrian Towse, “The R&D Cost of a New Medicine, 2012,” https://www.ohe.org/publications/rd-cost-new-medicine. The higher numbers are from Kyle Myers and Mark V. Pauly, “Pharma Trends Are Not What They Seem,” in Philip A. Rea, Mark V. Pauly, and Lawton R. Burns, Managing Discovery in the Life Sciences: Harnessing Creativity to Drive Biomedical Innovation, (Cambridge, U.K.: Cambridge University Press, 2018). In addition, the Tufts Center for the Study of Drug Development estimates the cost to be $2.6 billion. Tufts Center for the Study of Drug Development, “Cost to Develop and Win Marketing Approval for a New Drug is $2.6 Billion,” news release, November 18, 2014, http://csdd.tufts.edu/news/complete_story/pr_tufts_csdd_2014_cost_study. These numbers are in 2018 dollars.
[47]. Tufts Center for the Study of Drug Development, “Briefing: Cost of Developing a New Drug” (PowerPoint presentation, May 18, 2014).
[48]. John Vernon, Joseph Golec, and Joseph DiMasi, “Drug Development Costs When Financial Risk is Measured Using the Fama-French Three-Factor Model,” Health Economics 19, 4 (August 2010), https://www.ncbi.nlm.nih.gov/pubmed/19655335.
[49]. Jack Scannell, “Four Reasons Drugs Are Expensive, Of Which Two Are False,” Forbes, October 13, 2015, http://www.forbes.com/sites/matthewherper/2015/10/13/four-reasons-drugs-are-expensive-of-which-two-are-false/#257708d948a5.
[50]. Congressional Budget Office (CBO), “How Taxes Affect the Incentives to Invest in New Intangible Assets” (CBO, November 2018), 22–23, https://www.cbo.gov/system/files?file=2018-11/54648-Intangible_Assets.pdf.
[51]. PhRMA, “Chart Pack: Biopharmaceuticals in Perspective: Summer 2018,” 24–25.
[52]. Ibid, 25.
[53]. PhRMA, “Executive Summary: The Biopharmaceutical Pipeline,” 1.
[54]. TEConomy Partners, LLC, “The Economic Impact of the U.S. Biopharmaceutical Industry: National and State Estimates” (TEConomy Partners, May 2016), 1, http://phrma-docs.phrma.org/sites/default/files/pdf/biopharmaceuticaul-industry-economic-impact.pdf.
[55]. John Wu and Robert D. Atkinson, “How Technology-Based Start-Ups Promote U.S. Economic Growth” (Information Technology and Innovation Foundation, November 2017), https://itif.org/publications/2017/11/28/how-technology-based-start-ups-support-us-economic-growth. The life-sciences accounted for three of the ten industries that ITIF identified as being high-technology. The study defines start-ups as firms that are ten years old or less.
[56]. Ibid, 100–104.
[57]. Ibid, 11.
[58]. Joe Allen, “Compulsory Licensing for Medicare Drugs—Another Bad Idea from Capitol Hill,” IP Watchdog, August 23, 2018, http://www.ipwatchdog.com/2018/08/23/compulsory-licensing-medicare-drugs-bad-idea/id=100608/.
[59]. Association of University Technology Managers (AUTM), “AUTM 2017 Licensing Survey,” (AUTM, 2017), 5, https://autm.net/AUTM/media/SurveyReportsPDF/AUTM_2017_US_Licensing_Survey_no_appendix.pdf.
[60]. Evan Comen, “10 Universities Spending Billions on R&D,” MSN Money, April 4, 2017, https://www.msn.com/en-in/money/careersandeducation/10-universities-spending-billions-on-randd/ar-BBzjbN7?li=BBnb7Kz.
[61]. AUTM, “AUTM 2017 Licensing Survey.”
[62]. Robert Kneller, “The Importance of New Companies for Drug Discovery: Origins of a Decade of New Drugs,” Nature Reviews Drug Discovery 9 (November 2010): 867–882, https://www.ncbi.nlm.nih.gov/pubmed/21031002.
[63]. Ibid.
[64]. Bhaven N. Sampat and Frank R. Lichtenberg, “What Are The Respective Roles Of The Public And Private Sectors In Pharmaceutical Innovation?” Health Affairs 30, no. 2 (2011): 332–339, https://www.healthaffairs.org/doi/full/10.1377/hlthaff.2009.0917.
[65]. Derek Lowe, “Drugs Purely From Academia,” Science, February 2, 2016, https://blogs.sciencemag.org/pipeline/archives/2016/02/02/drugs-purely-from-academia.
[66]. Robert D. Atkinson, “How the Biopharmaceutical Industry Contributes to Open Scientific Knowledge” (Information Technology and Innovation Foundation, November 2018), 1–2, https://itif.org/publications/2018/11/05/how-biopharmaceutical-industry-contributes-open-scientific-knowledge.
[67]. “New Funding Awards—June 2018,” Duke University School of Medicine, July 2018, https://medicine.duke.edu/medicinenews/new-funding-awards-june-2018.
[68]. Brigid Sweeny, “In Era of Research Cuts, Romance Blossoming Much Earlier Between Universities and Big Pharma,” Crain’s Chicago Business, June 10, 2017, https://www.chicagobusiness.com/article/20170610/ISSUE01/170609858/big-pharmaceutical-companies-and-universities-partner-early.
[69]. Ibid.
[70]. Mark Zastrow, “The Top Academic and Corporate Partners in the Nature Index,” NatureINDEX, December 8, 2017, https://www.natureindex.com/news-blog/the-top-academic-and-corporate-partners-in-the-nature-index.
[71]. Alexander Schuhmacher et al., “Open Innovation and External Sources of Innovation. An Opportunity to Fuel the R&D Pipeline and Enhance Decision Making,” Journal of Translational Medicine (2018), https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-018-1499-2.
[72]. Ashley J. Stevens et al., “The Role of Public-Sector Research in the Discovery of Drugs and Vaccines,” The New England Journal of Medicine Vol. 364:6 (February 2011): 1, https://www.nejm.org/doi/full/10.1056/NEJMsa1008268.
[73]. Benjamin Zycher, Joseph A. DiMasi, and Christopher-Paul Milne, “The Truth About Drug Innovation: Thirty-Five Summary Case Histories on Private Sector Contributions to Pharmaceutical Science” (Center for Medical Progress at the Manhattan Institute, June 2008), 3–4, https://www.manhattan-institute.org/html/truth-about-drug-innovation-thirty-five-summary-case-histories-private-sector-contributions.
[74]. Ibid.
[75]. U.S. Senate Joint Economic Committee, “The Benefits of Medical Research and the Role of the NIH,” May 17, 2000, https://www.faseb.org/portals/2/pdfs/opa/2008/nih_research_benefits.pdf.
[76]. Organization for Economic Cooperation and Development (OECD), “Science, Technology and Innovation in the New Economy” (OECD: 2000), http://www.oecd.org/science/sci-tech/1918259.pdf.
[77]. “Breakthrough Business Models: Drug Development for Rare and Neglected Diseases and Individualized Therapies: Workshop Summary of Current Model for Financing Drug Development: From Concept Through Approval,” (Institute of Medicine (U.S.) Forum on Drug Discovery, Development, and Translation, Washington D.C., National Academies Press, 2009), 2, http://www.ncbi.nlm.nih.gov/books/NBK50972/.
[78]. “The Cost of Drug Development,” The New England Journal of Medicine, May 14, 2015, http://www.nejm.org/doi/full/10.1056/NEJMc1504317.
[79]. Battelle Technology Partnership Practice, “Patents as Proxies Revisited: NIH Innovation 2000 to 2013” (Battelle Technology Partnership Practice, 2015), https://www.acadrad.org/wp-content/uploads/2017/04/Patents-as-Proxies-Revisited.March_.Final_.pdf.
[80]. Azoulay, Li, Graff Zivin, and Sampat, “Public R&D Investments and Private-sector Patenting,” 3.
[81]. Battelle Technology Partnership Practice, “Patents as Proxies Revisited: NIH Innovation 2000 to 2013,” ES-1.
[82]. Iain M. Cockburn and Rebecca Henderson, “Publicly Funded Science and the Productivity of the Pharmaceutical Industry,” National Bureau of Economic Research Work Paper, No. 10775, 2001, http://www.nber.org/chapters/c10775.pdf.
[83]. Ibid.
[84]. Daniel Patrick Leyden and Albert N. Link, “Why Are Governmental R&D and Private R&D Complements,” Applied Economics 23 (1991): 23, https://www.tandfonline.com/doi/abs/10.1080/00036849100000132.
[85]. Ehrlich, “An Economic Engine,” 5.
[86]. Cockburn andHenderson, “Publicly Funded Science and the Productivity of the Pharmaceutical Industry,” 4.
[87]. Azoulay, Li, Graff Zivin, and Sampat, “Public R&D Investments and Private-sector Patenting,” 3, 22.
[88]. Ibid, 22.
[89]. Anusuya Chatterjee and Ross C. DeVol, “Estimating Long-Term Economic Returns of NIH Funding on Output in the Biosciences” (Milken Institute, 2012, 4), http://www.milkeninstitute.org/publications/view/535.
[90]. “The Impact of Genomics on the U.S. Economy” (Battelle Technology Partnership Practice, UMR, June 2013), http://www.unitedformedicalresearch.com/advocacy_reports/the-impact-of-genomics-on-the-u-s-economy.
[91]. Andrew A. Toole, “The Impact of Public Basic Research on Industrial Innovation: Evidence from the Pharmaceutical Industry,” Research Policy Vol. 41, Issue 1 (February 2012): 1–12, https://www.sciencedirect.com/science/article/abs/pii/S004873331100117X.
[92]. Frank R. Lichtenberg, “Pharmaceutical Innovation, Mortality Reduction, and Economic Growth,” NBER Working Paper No. 6569 (May 1998): 100–103, https://www.nber.org/papers/w6569.
[93]. Selma J. Mushkin and J. Steven Landefeld, Biomedical Research: Costs and Benefits (Cambridge, MA: Ballinger Publishing Co., 1979).
[94]. Sabarni K. Chatterjee and Mark L. Rohrbaugh, “NIH Inventions Translate Into Drugs and Biologics With High Public Health Impact,” Nature Biotechnology 32, (2014): 56, https://www.nature.com/articles/nbt.2785.
[95]. Robert A. Maxwell and Shohreh B. Eckhardt, Drug Discovery: A Case Book and Analysis (Clifton, New Jersey: Humana Press, 2011).
[96]. Stevens et al, “The Role of Public-Sector Research in the Discovery of Drugs and Vaccines.”
[97]. Ibid.
[98]. Thomas Sullivan, “NEJM: The Private Sector Discoveries Account for 79–90% of Pharmaceutical Products,” Policy & Medicine, May 5, 2018, https://www.policymed.com/2011/02/nejm-the-private-sector-discoveries-account-for-79-90-of-pharmaceutical-products.html.
[99]. Bhaven N. Sampat and Frank R. Lichtenberg, “What Are The Respective Roles Of The Public And Private Sectors In Pharmaceutical Innovation?” Health Affairs 30, No. 2 (2011): 332–339, https://www.healthaffairs.org/doi/full/10.1377/hlthaff.2009.0917.
[100]. Ibid, 336.
[101]. Zycher, DiMasi, and Milne, “The Truth About Drug Innovation,” 3.
[102]. Ibid.
[103]. Ibid, 29.
[104]. Ranjana Chakravarthy et al., “Public- and Private-Sector Contributions to the Research and Development of the Most Transformational Drugs in the Past 25 Years: From Theory to Therapy,” Therapeutic Innovation and Regulatory Science Vol. 50, 6 (July 2016), https://www.ncbi.nlm.nih.gov/pubmed/30231735.
[105]. Ibid.
[106]. Ibid.
[107]. Association of University Technology Managers (AUTM), “Activator Puts The Break on Cancer,” https://autm.net/about-tech-transfer/better-world-project/bwp-stories/activator-for-treating-cancer.
[108]. Ibid.
[109]. Ibid; Indranil Mallick, “The Roll of T-Cells in Cancer,” Verywellhealth, October 9, 2018, https://www.verywellhealth.com/t-cells-2252171.
[110]. AUTM, “Activator Puts The Break on Cancer.”
[111]. Ibid.
[112]. Ibid.
[113]. Robert Sanders, “UC Berkeley Research Led to Nobel Prize-winning Immunotherapy,” UC Berkeley news release, October 1, 2018, https://news.berkeley.edu/2018/10/01/uc-berkeley-research-led-to-nobel-prize-winning-immunotherapy/.
[114]. UC Berkeley Cancer Research Laboratory, “The Story of Yervoy/Ipilimumab,” http://crl.berkeley.edu/discoveries/the-story-of-yervoy-ipilimumab/.
[115]. “Blockade of T Lymphocyte Down-regulation Associated With CTLA-4 Signaling,” https://patents.google.com/patent/US5811097A/en.
[116]. Justia, “Blockade of T Lymphocyte Down-regulation Associated With CTLA-4 Signaling,” May 25, 2004, https://patents.justia.com/patent/20050249700/.
[117]. Bristol-Myers Squibb, “Bristol-Myers Squibb to Acquire Medarex,” news release, July 22, 2009, https://news.bms.com/press-release/partnering-news/bristol-myers-squibb-acquire-medarex.
[118]. UC Berkeley Cancer Research Laboratory, “The Story of Yervoy/Ipilimumab.”
[119]. AUTM, “Activator Puts The Break on Cancer.”
[120]. Sanders, “UC Berkeley Research Led to Nobel Prize-winning Immunotherapy.”
[121]. UC Berkeley Cancer Research Laboratory, “The Story of Yervoy/Ipilimumab.”
[122]. Ibid.
[123]. AUTM, “Activator Puts The Break on Cancer.”
[124]. Ibid.
[125]. Ibid.
[126]. Abbot, “Pulmonary Artery Pressure Monitoring,” https://www.cardiovascular.abbott/us/en/patients/living-with-your-device/heart-failure/pulmonary-pressure-artery-monitoring.html.
[127]. Brian Buntz, “How the CardioMEMS Device Was Designed,” Medical Device and Diagnostics Industry, August 26, 2015, https://www.mddionline.com/how-CardioMEMS-device-was-designed.
[128]. William T. Abraham et al., “Wireless Pulmonary Artery Haemodynamic Monitoring in Chronic Heart Failure: A Randomized Controlled Trial,” The Lancet, 377(9766) (February 19, 2011): 658-666, http://dx.doi.org/10.1016/S0140-6736(11)60101-3.
[129]. Buntz, “How the CardioMEMS Device Was Designed.”
[130]. Mitchel L. Zoler, “CardioMEMS Shows Real-world Success as Use Expands,” Cardiology News, October 17, 2017, https://www.mdedge.com/cardiology/article/149637/heart-failure/CardioMEMS-shows-real-world-success-use-expands.
[131]. Zoler, “CardioMEMS Shows Real-world Success as Use Expands.”
[132]. Buntz, “How the CardioMEMS Device Was Designed.”
[133]. Statement of Dr. Mark Allen, Joseph M. Pettit Professor and Regents Professor Georgia Institute of Technology, “Testimony to the House Committee on Science and Technology Subcommittee on Technology and Innovation,” July 17, 2007.
[134]. U.S. Patents 6,111,520 and 6,278,379; J.M. English and M.G. Allen, “Wireless Micromachined Ceramic Pressure Sensors,” Technical Digest, Twelfth IEEE International Conference on Micro Electro Mechanical Systems (1999): 511–516.
[135]. Allen, “Testimony to the House Committee on Science and Technology.”
[136]. Ibid.
[137]. Ibid.
[138]. Ibid.
[139]. Brad Perriello, “St. Jude Medical Closes $455M CardioMEMS Buy,” MassDevice, June 2, 2014, https://www.massdevice.com/st-jude-medical-closes-455m-CardioMEMS-buy/.
[140]. Jof Enriquez, “Abbott Completes $25 Billion Purchase Of St. Jude Medical,” MedDevice Online, January 6, 2017, https://www.meddeviceonline.com/doc/abbott-completes-billion-purchase-of-st-jude-medical-0001.
[141]. Philip A. Rea, Mark V. Pauly, and Lawton M. Burns, Managing Discovery in the Life Sciences: Harnessing Creativity to Drive Biomedical Innovation (Cambridge, England, Cambridge University Press: 2018), 333.
[142]. Ibid.
[143]. Ibid. 359.
[144]. National Cancer Institute, “How Imatinib Transformed Leukemia Treatment and Cancer Research,” https://www.cancer.gov/research/progress/discovery/gleevec.
[145]. National Cancer Institute, “How Imatinib Transformed Leukemia Treatment and Cancer Research.”
[146]. Philip A. Rea, Mark V. Pauly, and Lawton R. Burns, Managing Discovery in the Life Sciences, 343–346; Gerard Manning, “The Human Protein Kinases,” https://www.cellsignal.com/contents/science/protein-kinases-introduction/kinases.
[147]. Philip A. Rea, Mark V. Pauly, and Lawton R. Burns, Managing Discovery in the Life Sciences, 349.
[148]. Ibid., 349–350.
[149]. Ibid., 350–351.
[150]. Jessica Wapner, The Philadelphia Chromosome: A Mutant Gene and the Quest to Cure Cancer at the Genetic Level (New York: The Experiment, 2013).
[151]. Philip A. Rea, Mark V. Pauly, and Lawton R. Burns, Managing Discovery in the Life Sciences, 353–354.
[152]. Lawrence M. Fisher, “Post-Merger Integration: How Novartis Became No. 1,” Strategy + Business, April 1, 1998, https://www.strategy-business.com/article/16383?gko=28081.
[153]. National Cancer Institute, “How Imatinib Transformed Leukemia Treatment and Cancer Research.”
[154]. Ibid.
[155]. Philip A. Rea, Mark V. Pauly, and Lawton R. Burns, Managing Discovery in the Life Sciences, 356.
[156]. Penn Medicine, “FDA Approves Gene Therapy for Inherited Blindness Developed by the University of Pennsylvania and Children’s Hospital of Philadelphia,” news release, December 19, 2017, https://www.pennmedicine.org/news/news-releases/2017/december/fda-approves-gene-therapy-for-inherited-blindness-developed-by-university-of-pennsylvania-and-chop.
[157]. National Eye Institute, “NEI: 50 Years of Advances in Vision Research” (NEI, October 2018), 21, https://www.nei.nih.gov/sites/default/files/pdfs/NEI_Anniversary_History_Book.pdf.
[158]. NIH/National Eye Institute, “NIH Vision Researcher T. Michael Redmond Recognized with Champalimaud Vision Award,” EurekAlert, September 4, 2018, https://eurekalert.org/pub_releases/2018-09/nei-nvr083118.php.
[159]. Penn Medicine, “FDA Approves Gene Therapy for Inherited Blindness.”
[160]. Ibid. Penn colleagues Greg Acland, Tomas Aleman, Samuel Jacobson, and Artur Cideciyan also played pivotal roles, as did collaborations with Gus Aguirre (then at Cornell, now a professor of Medical Genetics and Ophthalmology at the University of Pennsylvania School of Veterinary Medicine) and Bill Hauswirth of the University of Florida.
[161]. National Eye Institute, “NEI: 50 Years of Advances in Vision Research.”
[162]. Children’s Hospital of Philadelphia, “Children With Congenital Blindness Can See After Gene Therapy,” news release, October 24, 2009, https://www.chop.edu/news/children-congenital-blindness-can-see-after-gene-therapy.
[163]. Penn Medicine, “FDA Approves Gene Therapy for Inherited Blindness.”
[164]. Spark Therapeutics, “The Development Timeline of Luxturna,” http://sparktx.com/wp-content/uploads/product-timeline.pdf.
[165]. Penn Medicine, “FDA Approves Gene Therapy for Inherited Blindness.”
[166]. Spark Therapeutics, “We Are Spark,” http://sparktx.com/we-are-spark/.
[167]. Chatterjee and Rohrbaugh, “NIH Inventions Translate Into Drugs and Biologics With High Public Health Impact.”
[168]. Stephen J. Ezell and Robert D. Atkinson, “ITIF Comments Responding to Administration Request for Information Regarding Federal Technology Transfer Authorities and Processes” (Information Technology and Innovation Foundation, August 2018), http://www2.itif.org/2018-nist-rfi-tech-transfer.pdf.
[169]. B. Graham, “Patent Bill Seeks Shift to Bolster Innovation,” The Washington Post, April 8, 1978; Stevens, “The Role of Public Sector Research,” 536.
[170]. Naomi Hausman, “University Innovation, Local Economic Growth, and Entrepreneurship,” U.S. Census Bureau Center for Economic Studies Paper No. CES-WP- 12–10 (July 2012): 5, https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2097842.
[171]. Louis G. Tornatzky and Elaine C. Rideout, “Innovation U 2.0: Reinventing University Roles in a Knowledge Economy” (State Science and Technology Institute, 2014) 165, https://ssti.org/report-archive/innovationu20.pdf.
[172]. Joseph P. Allen, “When Government Tried March In Rights to Control Health Care Costs,” IPWatchdog, May 2, 2016, http://www.ipwatchdog.com/2016/05/02/march-in-rights-health-care-costs/id=68816/.
[173]. Statement of Elmer B. Staats, comptroller general of the United States, Before the Committee on the Judiciary United States Senate, “S. 414-The University and Small Business Patent Procedures Act,” May 16, 1979, https://www.gao.gov/assets/100/99067.pdf.
[174]. Allen, “When Government Tried March In Rights to Control Health Care Costs.”
[175]. Ibid.
[176]. Ibid.
[177]. Hausman, “University Innovation, Local Economic Growth, and Entrepreneurship,” 6.
[178]. Joseph P. Allen, “A Reply to the New England Journal of Medicine,” IPWatchdog, September 10, 2013, https://www.ipwatchdog.com/2013/09/10/a-reply-to-the-new-england-journal-of-medicine/id=45085/.
[179]. Allen, “When Government Tried March In Rights to Control Health Care Costs.”
[180]. Ibid.
[181]. Ibid.
[182]. John H. Rabitschek and Norman J. Latker, “Reasonable Pricing—A New Twist for March-in Rights Under the Bayh-Dole Act,” Santa Clara High Technology Law Journal Vol. 22, Issue 1 (2005), 150, https://digitalcommons.law.scu.edu/cgi/viewcontent.cgi?article=1399&context=chtlj.
[183]. Joseph P. Allen, “Want a Greater ROI for Taxpayers? Restore the Patent System, Protect Bayh-Dole and Cut the Red Tape Strangling Federal Labs,” IPWatchdog, July 9, 2018, http://www.ipwatchdog.com/2018/07/09/roi-taxpayers-restore-the-patent-system/id=99136/.
[184]. David Mowery, “University-Industry Collaboration and Technology Transfer in Hong Kong and Knowledge-based Economic Growth,” (working paper, University of California, Berkeley, 2009), http://www.savantas.org/cmsimg/files/Research/HKIP/Report/1_mowery.pdf.
[185]. “Innovation’s Golden Goose,” The Economist, December 12, 2002, http://www.economist.com/node/1476653.
[186]. Wendy H. Schacht, “Patent Ownership and Federal Research and Development (R&D): A Discussion of the Bayh-Dole Act and the Stevenson-Wydler Act” (Congressional Research Service, 2000), http://ncseonline.org/nle/crsreports/science/st-66.cfm.
[187]. Hausman, “University Innovation, Local Economic Growth, and Entrepreneurship,” 7. (Calculation based on NBER patent data.)
[188]. Rabitschek and Latker, “Reasonable Pricing,” 150; The Economist, “Innovation’s Golden Goose.”
[189]. AUTM, “Driving the Innovation Economy: Academic Technology Transfer in Numbers,” https://www.autm.net/AUTMMain/media/SurveyReportsPDF/AUTM-FY2016-Infographic-WEB.pdf. Further, university research is about five times more likely to result in a licensed patented technology and about seven times more likely to result in an active patent license than research conducted at federal laboratories.
[190]. Dan Garcia, Duke University, Office of Licensing & Ventures, “The Bayh-Dole Act: Fueling Innovation for 38 Years,” news release, December 10, 2018, https://olv.duke.edu/news/bayh-dole-38-years/.
[191]. Lori Pressman et al., “The Economic Contribution of University/Nonprofit Inventions in the United States: 1996–2015” (Report prepared for AUTM and Biotechnology Industry Organization (BIO), June 2017), 3, https://www.bio.org/sites/default/files/June%202017%20Update%20of%20I-O%20%20Economic%20Impact%20Model.pdf.
[192]. AUTM, “Driving the Innovation Economy.”
[193]. AUTM, “FY2016 Licensing Survey,” https://www.autm.net/resources-surveys/research-reports-databases/licensing-surveys/fy2016-licensing-survey/.
[194]. Phone interview with Joe Allen, July 24, 2018.
[195]. Hausman, “University Innovation, Local Economic Growth, and Entrepreneurship,” 8.
[196]. Ibid.
[197]. Saul Lach and Mark Schankerman, “Incentives and Invention in Universities,” RAND Journal of Economics Vol 39, no. 2 (2008): 403–433, https://onlinelibrary.wiley.com/doi/pdf/10.1111/j.0741-6261.2008.00020.x.
[198]. Scott Shane, “Encouraging University Entrepreneurship? The Effect of the Bayh-Dole Act on University Patenting in the United States” Journal of Business Venturing 19 (2004): 127–151, https://papers.ssrn.com/sol3/papers.cfm?abstract_id=1495504.
[199]. Robert D. Atkinson, Stephen J. Ezell, and Luke A. Stewart, “The Global Innovation Policy Index” (Information Technology and Innovation Foundation and the Ewing Marion Kauffman Foundation, March 2012), 65, http://www2.itif.org/2012-global-innovation-policy-index.pdf.
[200]. California Senate Office of Research, “Optimizing Benefits From State-Funded Research” Policy Matters (March 2018), 12, https://sor.senate.ca.gov/sites/sor.senate.ca.gov/files/0842%20policy%20matters%20Research%2003.18%20Final.pdf.
[201]. Rabitschek and Latker, “Reasonable Pricing,” 150.
[202]. John R. Thomas, “March-In Rights Under the Bayh-Dole Act” (Congressional Research Service, August 2016), 7, https://fas.org/sgp/crs/misc/R44597.pdf.
[203]. David S. Bloch, “Alternatives to March-In Rights” Vanderbilt Journal of Entertainment and Technology Law, Vol. 18:2:247 (2016): 253, http://www.jetlaw.org/wp-content/uploads/2016/03/Bloch_SPE_6-FINAL.pdf.
[204]. Statement of Senator Birch Bayh to the National Institutes of Health (May 24, 2004), https://www.ott.nih.gov/sites/default/files/documents/2004NorvirMtg/2004NorvirMtg.pdf.
[205]. Thomas, “March-In Rights Under the Bayh-Dole Act,” 11.
[206]. Ibid.
[207]. Ibid, 14.
[208]. Joseph Allen, “Proposal from Senator King Won’t Reduce Drug Prices, Just Innovation,” IPWatchdog, July 17, 2017, https://www.ipwatchdog.com/2017/07/17/senator-king-reduce-drug-prices-innovation/id=85702/.
[209]. Thomas, “March-In Rights Under the Bayh-Dole Act,” 10.
[210]. Birch Bayh, “Statement of Birch Bayh to the National Institutes of Health,” May 25, 2014, http://www.essentialinventions.org/drug/nih05252004/birchbayh.pdf.
[211]. Birch Bayh and Bob Dole, “Our Law Helps Patients Get New Drugs Sooner,” The Washington Post, April 11, 2002, https://www.washingtonpost.com/archive/opinions/2002/04/11/our-law-helps-patients-get-new-drugs-sooner/d814d22a-6e63-4f06-8da3-d9698552fa24/.
[212]. Ibid.
[213]. Allen, “When Government Tried March In Rights to Control Health Care Costs.”
[214]. National Institute of Standards and Technology (NIST), “Return on Investment Initiative: Draft Green Paper” (NIST, December 2018), 30, https://nvlpubs.nist.gov/nistpubs/SpecialPublications/NIST.SP.1234.pdf.
[215]. Ibid, 30.
[216]. Thomas, “March-In Rights Under the Bayh-Dole Act,” 9.
[217]. Ibid.
[218]. Ibid.
[219]. Thomas, “March-In Rights Under the Bayh-Dole Act,” 11–12.
[220]. Elias A. Zerhouni, director, NIH, “In the Case of Norvir Manufactured by Abbott Laboratories, Inc.,” July 29, 2004, http://www.ott.nih.gov/sites/default/files/documents/policy/March-In-Norvir.pdf.
[221]. Rabitschek and Latker, “Reasonable Pricing,” 160.
[222]. Ibid, 167.
[223]. Peter Amo and Michael Davis, “Why Don’t We Enforce Existing Drug Price Controls? The Unrecognized and Unenforced Reasonable Pricing Requirements Imposed upon Patents Deriving in Whole or in Part from Federally Funded Research,” Tulane Law Review Vol. 75, 631 (2001), https://engagedscholarship.csuohio.edu/cgi/viewcontent.cgi?article=1754&context=fac_articles.
[224]. Ibid.
[225]. Rabitschek and Latker, “Reasonable Pricing,” 162–163.
[226]. Ibid, 163.
[227]. Ibid. Here, the Presidential Memoranda refers to memoranda produced by the Kennedy and Nixon administrations that pertained to government policy related to contractor ownership of inventions.
[228]. Ibid.
[229]. Stephen Ezell interview with Joseph P. Allen, November 18, 2018.
[230]. National Institutes of Health, “NIH News Release Rescinding Reasonable Pricing Clause” news release, April 11, 1995, https://www.ott.nih.gov/sites/default/files/documents/pdfs/NIH-Notice-Rescinding-Reasonable-Pricing-Clause.pdf.
[231]. Ibid.
[232]. Allen, “Compulsory Licensing for Medicare Drugs—Another Bad Idea from Capitol Hill.”
[233]. National Institutes of Health, “OTT Statistics,” accessed January 13, 2019, https://www.ott.nih.gov/reportsstats/ott-statistics.
[234]. National Institutes of Health, “NIH News Release Rescinding Reasonable Pricing Clause.”
[235]. Allen, “Compulsory Licensing for Medicare Drugs—Another Bad Idea from Capitol Hill.”
[236]. California Senate Office of Research, “Optimizing Benefits From State-Funded Research,” 11.
[237]. Eric Sagonowsky, “Sanofi Pulls Out of Zika Vaccine Collaboration as Feds Gut its R&D Contract,” FiercePhrma, September 1, 2017, https://www.fiercepharma.com/vaccines/contract-revamp-sanofi-s-Zika-collab-u-s-government-to-wind-down; Ed Silverman, “Sanofi Rejects U.S. Army Request for ‘Fair” Pricing’ for a Zika Vaccine,” PBS, May 20, 2017, https://www.pbs.org/newshour/health/sanofi-army-request-pricing-Zika-vaccine.
[238]. Bernie Sanders, “Trump Should Avoid a Bad Zika Deal,” The New York Times, March 10, 2017, https://www.nytimes.com/2017/03/10/opinion/bernie-sanders-trump-should-avoid-a-bad-Zika-deal.html.
[239]. Eric Sagonowsky, “Sanofi Executive Lays Out Case For Taxpayer Funding—And Exclusive Licensing—on Zika Vaccine R&D,” FiercePhrma, May 25, 2017, https://www.fiercepharma.com/vaccines/exec-says-corporate-responsibility-not-potential-profits-drove-sanofi-s-Zika-vaccine-r-d.
[240]. Jeannie Baumann, “Sanofi Never Rejected Fair Pricing for Zika Vaccine: Exec,”BloombergNews, July 18, 2017, https://www.bna.com/sanofi-rejected-fair-n73014461930/.
[241]. Sagonowsky, “Sanofi Executive Lays Out Case For Taxpayer Funding—And Exclusive Licensing—on Zika Vaccine R&D.”
[242]. Sagonowsky, “Sanofi Pulls Out of Zika Vaccine Collaboration as Feds Gut its R&D Contract.”
[243]. Joseph Allen, “Bayh-Dole Under March-in Assault: Can It Hold Out?” IP Watchdog, January 21, 2016, http://www.ipwatchdog.com/2016/01/21/bayh-dole-under-march-in-assault-can-it-hold-out/id=65118/.
[244]. AUTM, “Driving the Innovation Economy.”
[245]. Stephen J. Ezell, “Why Exploiting Bayh-Dole ‘March-In’ Provisions Would Harm Medical Discovery,” April 29, 2016, https://www.innovationfiles.org/why-exploiting-bayh-dole-march-in-provisions-would-harm-medical-discovery/.
[246]. Bloch, “Alternatives to March-In Rights,” 261.
[247]. Ibid.
[248]. NIST, “Return on Investment Initiative: Draft Green Paper,” 31.
[249]. Ibid, 33.
[250]. NIST, “Return on Investment Initiative: Draft Green Paper,” 26.
[251]. Ibid., 28; U.S. Government Accountability Office (GAO), “Agencies’ Rights to Federally Sponsored Biomedical Inventions” (GAO, 2003), https://www.gao.gov/new.items/d03536.pdf.
[252]. NIST, “Return on Investment Initiative: Draft Green Paper,” 28.
[253]. TEConomy Partners, LLC, “Closing the Gap: Increasing Global Competition to Attract and Grow the Biopharmaceutical Sector,” (TEConomy Partners, LLC, June 2017), iv, https://www.phrma.org/report/closing-the-gap-increasing-global-competition-to-attract-and-grow-the-biopharmaceutical-sector.
[254]. Ibid, 14.
[255]. OECD, “Government Budget Appropriations or Outlays for R&D,” (accessed February 17, 2019), Dhttps://stats.oecd.org/Index.aspx?DataSetCode=GBAORD_NABS2007#.
[256]. “China’s Biotech Sector to Exceed 4% of GDP by 2020: Authority,” ChinaDaily, May 3, 2017, http://www.chinadaily.com.cn/business/2017-05/03/content_29181073.htm.
[257]. Megan Molteni, “A Chinese Genome Giant Sets Its Sights on the Ultimate Sequencer,” Wired, May 18, 2017, https://www.wired.com/2017/05/chinese-genome-giant-sets-sights-uitimate-sequencer/.
